
カテゴリー: バイオインフォマティクス
脊椎動物最大ゲノムのハイギョのRNA-seq解析にはどの程度のスペックのパソコンが必要か?
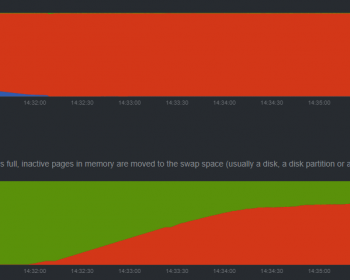
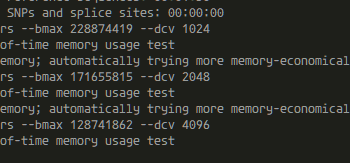
タイトルの通り。これからハイギョのRNA-seq解析などをしたいという稀有な研究者に向けたメモ。 BLAST 正直BLASTするだけならそこまでスペックはいらない。他のゲノムと比べると時間がかかるが、数コアでも十分可能。 STAR を用いた RNA-seq マッピング RNA-seqでもTrinit…
LASTZで全ゲノムをアライメント

2022/06/30 Seqkit のソート項目を追記2025/02/04 最新版へのリンクへ修正 lastzについての日本語ドキュメントが少ないので備忘録として。 lastzはWhole genomeレベルでのアライメントができるツールである。 結構古くからあり有名なのでこの辺の説明は割愛。今も開…
TE配列の分類を行うDeepTEの使い方

最近は転移因子(TE、トランスポゾン)の話が連日続いているが、今回は前回の記事で紹介した機械学習によるTE分類ツールの1つ、DeepTE (Yan et al., 2020) を実際に使ってみる。 Yan, H., Bombarely, A., & Li, S. (2020). D…
転移因子(TE)の分類ツール: ClassifyTE、DeepTE

転移因子(トランスポゾン、TE)はゲノムのある位置からある位置へと飛んでいくというDNA配列である。様々な真核生物においてゲノムに存在し、様々な役割を担っていたり、あるいは単純に「ジャンクDNA」であるようなものも存在する(最初はジャンクとして見られていたけど近年になって様々な役割が明らかにされてき…
ゲノム中の反復配列を検出するソフト、REpeat Detector : Redの使い方

ゲノム中の反復配列、トランスポゾン等を検出するソフトは色々と開発されている。代表的なものにRepeat Maskerだったり、RepeatScout、WindowMasker、RepeatModeler等々ある。 それぞれライブラリをベースに検出を行ったり、あるいは denovo 法に基づいていたり…
枝が長い系統樹の注意点 – Long Branch Attraction
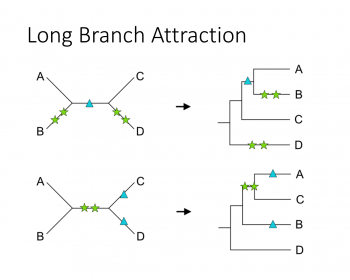
分子系統樹を作成する時、特に何も考えずにツールに突っ込んで得られたデータの樹形を見ているということはないだろうか? 系統樹に関する既知の問題点として「Long branch attraction」という現象が古くから(50年くらい前)知られている。ロングブランチアトラクション、の日本語訳はまだ当てら…
NovaseqやNextSeqのシーケンスデータにポリG配列(poly-G)が含まれる
先日またRNA-seqを外注した。 FASTQCでクォリティチェックを行ったところ、以下のように、”Overrepresented sequences”に関する表示が出ていた。なにやら”GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG…
Featurecountsで”Successfully assigned alignments : 0 (0.0%)”と出る時

FeatureCountsを使ってマッピングしたRNA-seqリードをカウントしようとした時、どうしても全てが「0」カウントで出力されることがあった。 BAMファイルを調べる。 すると、GFFファイルとはぜんぜん異なる染色体の番号が……よくよく元のSTARのパラメータを記したshファイルを読み返すと…
EnTAPでde novoアセンブルしたRNA-seqデータにアノテーション – 実データ編

前回のインストールの記事からめちゃくちゃ時間が経ってしまった。サクッと続きとして実データでの稼働方法を記しておく。 このドキュメントをベースに。 Trinityでde novo assemble Trinityで最初に自分のRNA-seqデータをアセンブルしておく。Trinity以外でも別に問題はな…
脊椎動物最大ゲノムのハイギョのRNA-seq解析にはどの程度のスペックのパソコンが必要か?
タイトルの通り。これからハイギョのRNA-seq解析などをしたいという稀有な研究者に向けたメモ。 BLAST 正直BLASTするだけならそこまでスペックはいらない。他のゲノムと比べると時間がかかるが、数コアでも十分可能。 STAR を用いた RNA-seq マッピング RNA-seqでもTrinit…
LASTZで全ゲノムをアライメント
2022/06/30 Seqkit のソート項目を追記2025/02/04 最新版へのリンクへ修正 lastzについての日本語ドキュメントが少ないので備忘録として。 lastzはWhole genomeレベルでのアライメントができるツールである。 結構古くからあり有名なのでこの辺の説明は割愛。今も開…
TE配列の分類を行うDeepTEの使い方
最近は転移因子(TE、トランスポゾン)の話が連日続いているが、今回は前回の記事で紹介した機械学習によるTE分類ツールの1つ、DeepTE (Yan et al., 2020) を実際に使ってみる。 Yan, H., Bombarely, A., & Li, S. (2020). D…
転移因子(TE)の分類ツール: ClassifyTE、DeepTE
転移因子(トランスポゾン、TE)はゲノムのある位置からある位置へと飛んでいくというDNA配列である。様々な真核生物においてゲノムに存在し、様々な役割を担っていたり、あるいは単純に「ジャンクDNA」であるようなものも存在する(最初はジャンクとして見られていたけど近年になって様々な役割が明らかにされてき…
ゲノム中の反復配列を検出するソフト、REpeat Detector : Redの使い方
ゲノム中の反復配列、トランスポゾン等を検出するソフトは色々と開発されている。代表的なものにRepeat Maskerだったり、RepeatScout、WindowMasker、RepeatModeler等々ある。 それぞれライブラリをベースに検出を行ったり、あるいは denovo 法に基づいていたり…
枝が長い系統樹の注意点 – Long Branch Attraction
分子系統樹を作成する時、特に何も考えずにツールに突っ込んで得られたデータの樹形を見ているということはないだろうか? 系統樹に関する既知の問題点として「Long branch attraction」という現象が古くから(50年くらい前)知られている。ロングブランチアトラクション、の日本語訳はまだ当てら…
NovaseqやNextSeqのシーケンスデータにポリG配列(poly-G)が含まれる
先日またRNA-seqを外注した。 FASTQCでクォリティチェックを行ったところ、以下のように、”Overrepresented sequences”に関する表示が出ていた。なにやら”GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG…
Featurecountsで”Successfully assigned alignments : 0 (0.0%)”と出る時
FeatureCountsを使ってマッピングしたRNA-seqリードをカウントしようとした時、どうしても全てが「0」カウントで出力されることがあった。 BAMファイルを調べる。 すると、GFFファイルとはぜんぜん異なる染色体の番号が……よくよく元のSTARのパラメータを記したshファイルを読み返すと…
EnTAPでde novoアセンブルしたRNA-seqデータにアノテーション – 実データ編
前回のインストールの記事からめちゃくちゃ時間が経ってしまった。サクッと続きとして実データでの稼働方法を記しておく。 このドキュメントをベースに。 Trinityでde novo assemble Trinityで最初に自分のRNA-seqデータをアセンブルしておく。Trinity以外でも別に問題はな…





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)

