Cell Hashing による複数サンプルデータ読み込み

今回はシングルセルシーケンスにおける Multiplex Samples の取り扱いについて簡単に書き残しておく。 昨今流行しているシングルセルシーケンスであるが、複数のサンプルにそれぞれ “Hashtag” (ハッシュタグ) をつけることで、混合してシングルセルシーケンスし…

今回はシングルセルシーケンスにおける Multiplex Samples の取り扱いについて簡単に書き残しておく。 昨今流行しているシングルセルシーケンスであるが、複数のサンプルにそれぞれ “Hashtag” (ハッシュタグ) をつけることで、混合してシングルセルシーケンスし…
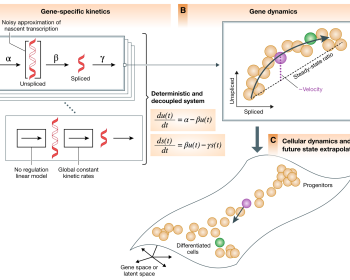
今回は RNA velocity (直訳するとRNA速度だが、適切な語訳ではないため、RNA velocityとして記述する)について説明する。 RNA velocity の基礎知識 RNA velocity は2018年に La Manno らが発表・提唱したもので、(一般に)scRNA-seq …

ハイギョは現生魚類の中で最も陸上脊椎動物(両生類や有羊膜類など)に近い生物である。その名の通り肺を持っている魚である。エラも持っており、エラと肺、両方で呼吸ができる特殊な魚である(他にもポリプテルスなどが同様に肺とエラ両方持っている)。 また、一部のハイギョでは水が干上がった乾季などにおいては泥…
タイトルの通り。これからハイギョのRNA-seq解析などをしたいという稀有な研究者に向けたメモ。 BLAST 正直BLASTするだけならそこまでスペックはいらない。他のゲノムと比べると時間がかかるが、数コアでも十分可能。 STAR を用いた RNA-seq マッピング RNA-seqでもTrinit…

先日またRNA-seqを外注した。 FASTQCでクォリティチェックを行ったところ、以下のように、”Overrepresented sequences”に関する表示が出ていた。なにやら”GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG…

FeatureCountsを使ってマッピングしたRNA-seqリードをカウントしようとした時、どうしても全てが「0」カウントで出力されることがあった。 BAMファイルを調べる。 すると、GFFファイルとはぜんぜん異なる染色体の番号が……よくよく元のSTARのパラメータを記したshファイルを読み返すと…

前回のインストールの記事からめちゃくちゃ時間が経ってしまった。サクッと続きとして実データでの稼働方法を記しておく。 このドキュメントをベースに。 Trinityでde novo assemble Trinityで最初に自分のRNA-seqデータをアセンブルしておく。Trinity以外でも別に問題はな…
2021/05/25 追記: Ubuntuでのインストール方法を更新 前回の話とのつながりとしては、STARでRNA-seqのリードをリファレンスゲノムにマッピングしたという前提である。 さて、このマッピングしただけのデータ(SAMファイル)では統計的な解析に向かないという問題点がある。もちろん、こ…

最近更新を続けているEnTAPでde novo assemble するRNA-seqデータにアノテーションをつけるプロジェクトの下準備編。 今回はTrinityでde novo アセンブルを行う。 2020/07/09 追記: 出力結果の見方を追記 目的と実行環境 複数のRNA-seqデータをマッピ…
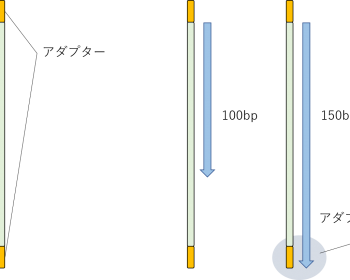
2022/06/02 Trimmomaticの記述を削除、代わりにfastpを追加。 今回はマクロジェンに委託したRNA-seqについての話。弊ラボではそこにRNA-seqを委託しているので、ラボメンに向けての備忘録も兼ねて。 シーケンスの状態 弊研究室ではRNAシーケンスをマクロジェンに委託してい…





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)

