
2022/06/02 Trimmomaticの記述を削除、代わりにfastpを追加。
今回はマクロジェンに委託したRNA-seqについての話。弊ラボではそこにRNA-seqを委託しているので、ラボメンに向けての備忘録も兼ねて。
シーケンスの状態
弊研究室ではRNAシーケンスをマクロジェンに委託している。シーケンサはNovaSeq6000で100bpもしくは150bpで読んでいる。相乗りで委託することで返ってくるまでの時間はかかるが、比較的安価に委託できる。質については他と比較したことがないのでわからないが、よく読めていると思うし、クォリティチェックもしっかりやってくれるので安心である。
ただ、100bpで委託したときはQC(クォリティチェック)のときに特別問題はなかったが、150bpで委託したときにはアダプター配列が含まれていると出た。仕様書を見るとどちらも「アダプター配列は除去していない」と書かれているので、本来は含まれるものなのであろう。100bpと150bpで含まれる・含まれない理由をマクロジェン側に尋ねると、
今回の条件では、アダプターの両側から、シーケンスプライマー直下の
150ntのシーケンスを行っております。
そのため、インサートサイズが150bpに満たない場合は、
反対側のアダプター配列がシーケンスされることとなります。
インサートサイズが150bpを越える場合は、反対側のアダプター配列に
到達しないため、アダプター配列がリード情報に含まれなくなります。
ということらしい。わかりやすく図説するとこのようになる。と思う。
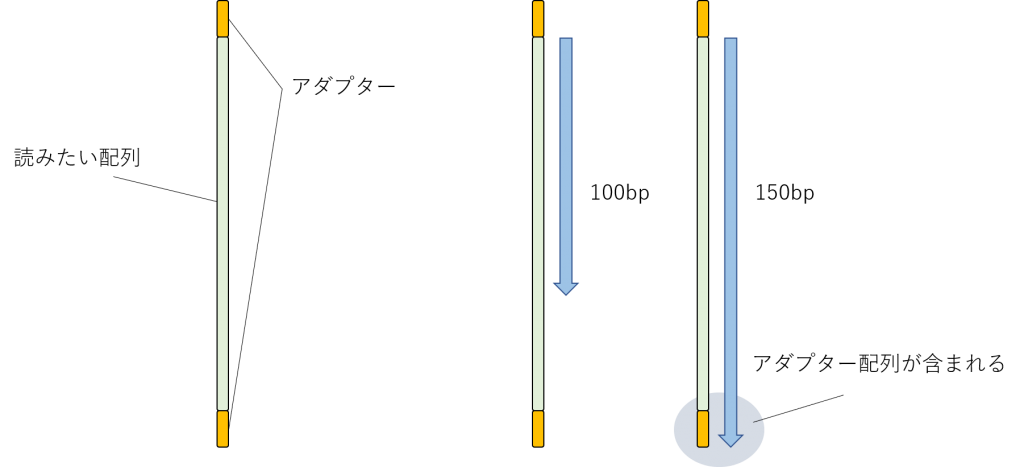
この読みたい配列(DNA Insert)の長さはまちまちなので、長くシーケンスするとアダプター配列まで読まれてしまうものが出てくるということらしい。
なるほど。
アダプター配列を取り除く
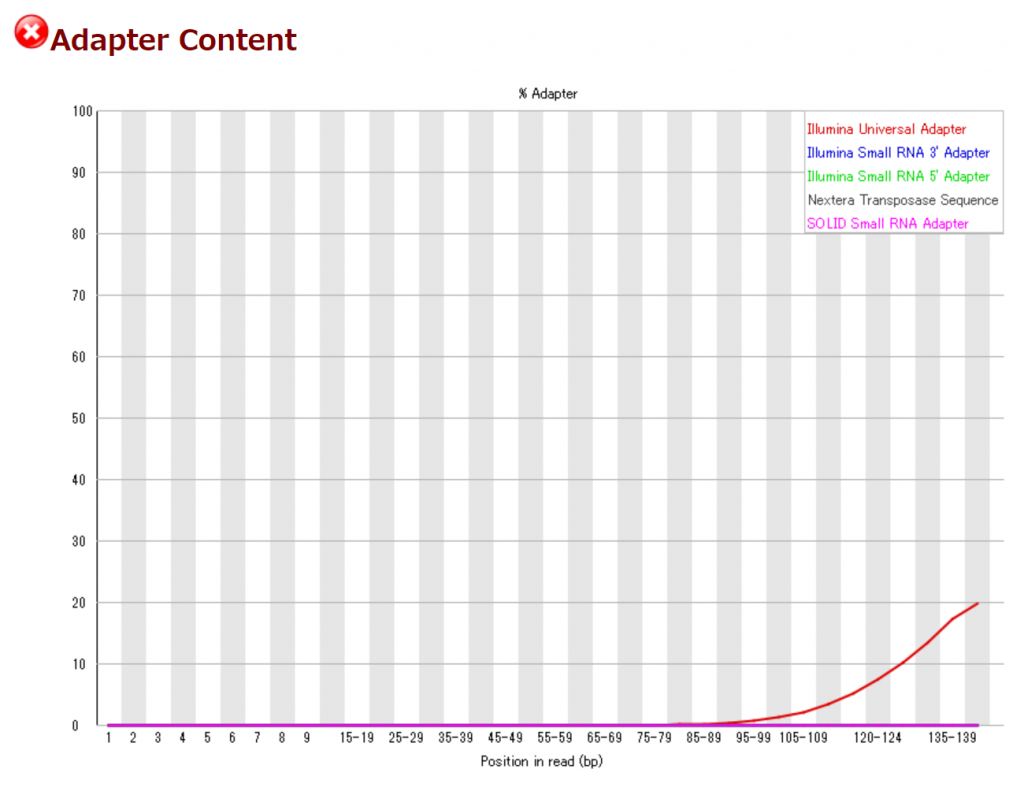
で、「このアダプター配列の何が問題なの?」ってことだが、「マッピング率がすこぶる悪くなる」ってことだ。今回100bpやっていて、追加で150bpの長さで読んでもらったので気づけたが、明らかにマッピング率が悪くなる。
100bp長のときは90%あったマッピング率が150bp長でシーケンスしたときのをマッピングすると65%にまで落ち込んだ。はて。と思って今回のこれにつながったわけだが、何はともあれアダプター配列を取り除いてやらねばならない。
Quality Control のためのソフトウエアである“prinseq++”を用いて無理矢理その部分を一括60bp削ってやるとかいう荒業でとりあえずやってみたところ、マッピング率は90%弱まで回復した。
まあでもそんなことしてしまったら大事な配列の部分まで失ってしまうのでアダプター配列だけを取り除きたい。
(2022/06/02 修正。Trimmomaticを紹介していたが、全然トリミングできていなかった。私自身とっくに使っていなかったが指摘があったので修正。ありがとうございます)
そこでfastpというツールを使う。
Chen, S., Zhou, Y., Chen, Y., & Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 34(17), i884-i890.
詳しい解説は別記事にあるが、特にオプションを指定しなくともデフォルトでアダプターを検知、削除をしてくれる。その他、ポリG配列なども削除してくれる。
その結果を再びFastQCにかけて比較
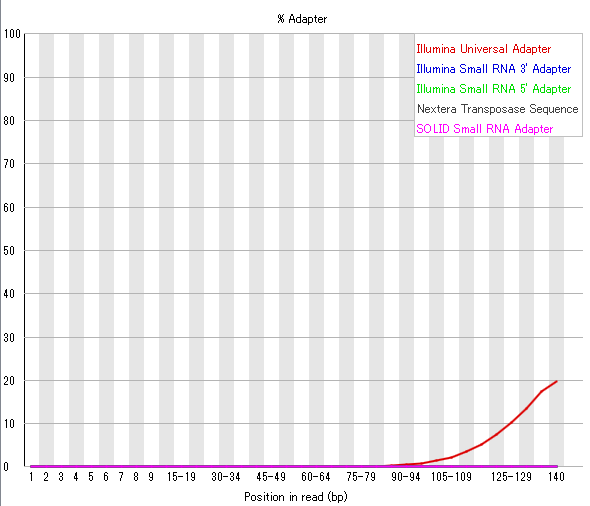
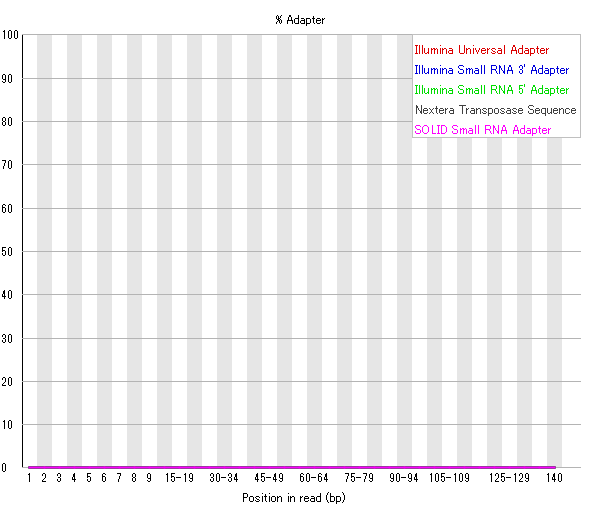
Trimming前のものと比べれば一目瞭然で良くなっていることがわかる。とりあえず使えそうだ。
おわりに
今回はシーケンスの結果からアダプター配列を取り除く重要性について簡単に述べた。ただ送られてきた結果を脳死でパイプラインに突っ込む前にもう少しそのRAWデータを精査しておく必要があることが伝わっただろうか。
この操作も後々の解析結果に大きく響いてくるのでよく調整する必要があるところである。





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)

