ScipyでDunnettの検定をする

Scipyの最新バージョン(1.12.0)ではDunnettの検定が実装されている。Dunnett検定については別途調べてほしいが、多重検定のひとつだ。コントロールとなる群といくつかの実験群を比較し、検定する。 Scipyを使えば簡単にできる。以下は公式ドキュメントのサンプルの簡単な翻訳。 この…

Scipyの最新バージョン(1.12.0)ではDunnettの検定が実装されている。Dunnett検定については別途調べてほしいが、多重検定のひとつだ。コントロールとなる群といくつかの実験群を比較し、検定する。 Scipyを使えば簡単にできる。以下は公式ドキュメントのサンプルの簡単な翻訳。 この…
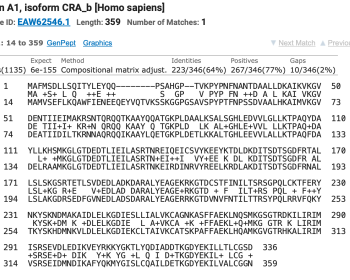
tblastn、blastp をして結果を眺めて整理していたとき、あまり気にしていなかったが “Positives” というスコアがあることに気づいた。 そのとなりにある “Identities” はアライメントされたアミノ酸残基の完全一致率、数を示し…

タンパク質のRefSeqID(NPやXPから始まるID)を大量に持っていて、それをPython上でなんとかGene symbolに変換できないかを模索していた。 そんなとき、MyGene.info というサービスを見つけた。あまり日本語のドキュメントがないので軽く紹介しておく。 MyGene.i…

ハイギョは現生魚類の中で最も陸上脊椎動物(両生類や有羊膜類など)に近い生物である。その名の通り肺を持っている魚である。エラも持っており、エラと肺、両方で呼吸ができる特殊な魚である(他にもポリプテルスなどが同様に肺とエラ両方持っている)。 また、一部のハイギョでは水が干上がった乾季などにおいては泥…
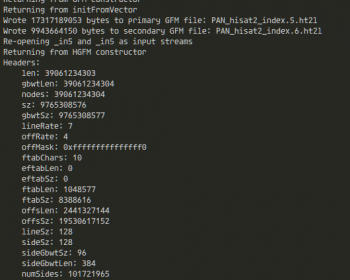
遺伝研スパコン(遺伝研スーパーコンピュータシステム)では、Grid Engine (SGE系)を採用している。この記事を読んでいる人(利用者)であれば、まず最初にqloginしろ、とか、ジョブを実行するときはスクリプトファイルをqsubしろ、とかを講習会などで最初に習うはずである。 無事終了する…
イントロンの解析をする際に、既存のGFF/GTFのアノテーションファイルにIntron情報が入っていない場合がある。そうしたときにちまちまエキソン情報から計算してもいいが、便利なツールがいくつかあるので紹介する。 AGATを使う これが現状一番オススメの方法。GFF/GTFの変換のために使ったが…
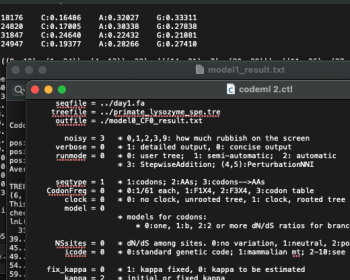
選択圧(淘汰圧)を解析するためにPAMLのcodemlがよく使われる。 いくつか日本語でも解説が書かれており、それに従って使っていることが多かったが、CodonFrequencyについての記述は日英共に解説が少ない。どうやって選択すべきか調べていたが、灯台もと暗しで知人がきちんと統計的なモデル選択の…
NCBIのAPIでFASTAなどを取得するツールを作っているのだが、非同期処理を導入して大量に並列取得をしようとしたところ”API rate limit exceeded”とエラーが出ていることが判明した。 ググると以下のページのように、アクセス制限が存在することが明らかにな…
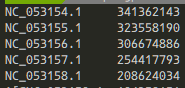
参考: Sequence length from Fasta マルチFASTA で、それぞれの配列の長さを出してほしい時がある。 そういったときに簡単に出力してくれるのがbioawkだ。 で、仮想環境上にインストール。 で、出力してくれる。 全部出すと長い場合、パイプで繋いでやって欲しい部分に絞って…
バイオインフォマティクス関連で、稀にtwoBit file (2bit file) を使用する機会がある。 通常、ゲノムファイルなどのシーケンスデータはFASTA形式で頒布されているが、一部において効率的・高速な解析のためにtwoBit fileと呼ばれる形式が使用される。 UCSCによると、「ゲノ…





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)

