遺伝研スパコン(Linux)でリファレンスゲノムを集める

遺伝研スパコンやLinux, Ubuntuなどの環境でNCBIのリファレンスゲノムを集める方法を基本から抑える。学部生に指導したときの資料を下敷きにしている。 ゲノムの選定方法などについては以前の記事を参照してほしい。 Linux基本コマンド 簡単におさらいしておく。 カレントディレクトリの確…

遺伝研スパコンやLinux, Ubuntuなどの環境でNCBIのリファレンスゲノムを集める方法を基本から抑える。学部生に指導したときの資料を下敷きにしている。 ゲノムの選定方法などについては以前の記事を参照してほしい。 Linux基本コマンド 簡単におさらいしておく。 カレントディレクトリの確…

今回からの記事は IBM の Qiskit で量子機械学習をマルチオミクスデータに向けてやろうというものである。昨年のカンファレンスの資料と Notebook が公開されていてダウンロードしていたのだが、論文やらなんやらで色々忙しいのを言い訳に塩漬けにしていたのをなんとか掘り起こそうという備忘録だ。…
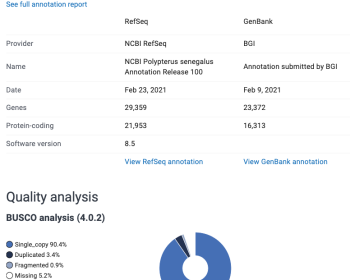
導入 ヒトやマウスといったモデル生物以外のゲノムが染色体レベルで組み上がる時代に突入している。ゲノム「解読」黎明期ではゲノムを決定することで様々な生命現象が明らかになると思われていたが、実際は一次元の塩基配列だけでわかることには限界があるということがその後段々と浸透していった。そのため現在では「ゲノ…

ヒトやマウスなどのモデル生物ではRepbaseなどのリピート配列やTE(トランスポゾン)配列などのアノテーションが充実している。一方で、新規にゲノムを決定する・した生物ではそれがそもそもされていない場合が多い。 de novoでライブラリを作成し、アノテーションするパイプラインのゴールドスタンダード…
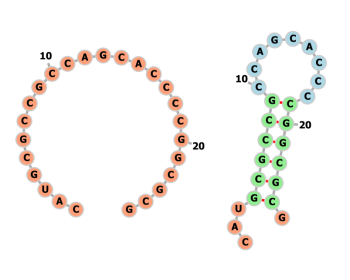
近年量子コンピュータが注目されているが、応用先については「創薬」「金融」といった抽象的なものはよく取り上げられるが実際どの程度の具体的な事例に適用できるかについてはあまり耳にしないと思う。 今回はIBMとモデルナ(新型コロナウイルスのmRNAワクチンで有名になった会社)が実際に協働して量子コンピュー…
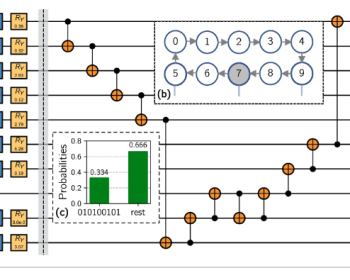
今回は量子コンピュータが生命科学にどういう形でアプローチできるのか、最近(2022年)のレビューを通じて紹介したい。量子コンピュータに限らず、現在の動向を色々と抑えて書き留めたいと思う。 最近の計算生命科学(バイオインフォ)の動向 最近のバイオインフォマティクスの分野の発展は著しい。特にタンパク質の…
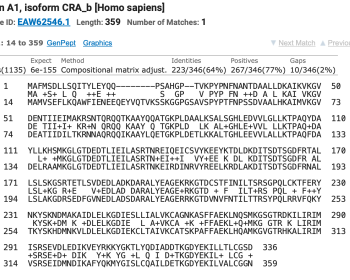
tblastn、blastp をして結果を眺めて整理していたとき、あまり気にしていなかったが “Positives” というスコアがあることに気づいた。 そのとなりにある “Identities” はアライメントされたアミノ酸残基の完全一致率、数を示し…

タンパク質のRefSeqID(NPやXPから始まるID)を大量に持っていて、それをPython上でなんとかGene symbolに変換できないかを模索していた。 そんなとき、MyGene.info というサービスを見つけた。あまり日本語のドキュメントがないので軽く紹介しておく。 MyGene.i…

ハイギョは現生魚類の中で最も陸上脊椎動物(両生類や有羊膜類など)に近い生物である。その名の通り肺を持っている魚である。エラも持っており、エラと肺、両方で呼吸ができる特殊な魚である(他にもポリプテルスなどが同様に肺とエラ両方持っている)。 また、一部のハイギョでは水が干上がった乾季などにおいては泥…
イントロンの解析をする際に、既存のGFF/GTFのアノテーションファイルにIntron情報が入っていない場合がある。そうしたときにちまちまエキソン情報から計算してもいいが、便利なツールがいくつかあるので紹介する。 AGATを使う これが現状一番オススメの方法。GFF/GTFの変換のために使ったが…





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)

