
私はRNA-seqのリードのマッピングにSTARを使用しているのだが、ラボの私以外のメンバーはHISAT2を使用している。なんかHISAT2を使用している先輩の結果が「マッピング率が低い」という話だったので実際どの程度違いものなのか気になり、今回調べてまとめることとした。
RNA-seqのマッピングツール
マッピングに用いるソフトウエアは様々なものが公開されている。有名所でいうとHISAT2が挙げられるだろう。ただ現在においてもTopHatなどの古いマッピングツールが用いられていることがあり、再現性などの点において問題視されている(Simoneau et al., 2019)。
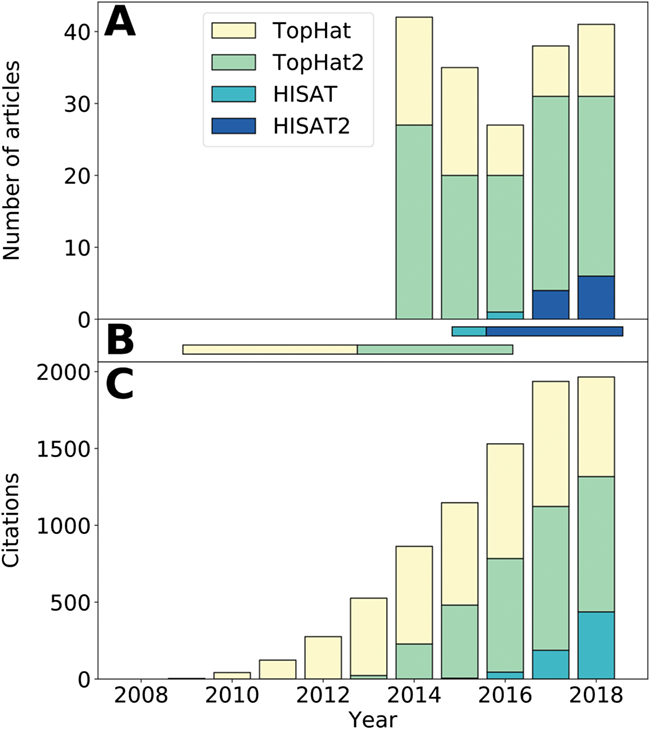
示した図。 Simoneau et al., 2019 より引用 改変なし。
図1Bにどのソフトウエアが”推奨”されてきたかが記載されている通り、現在はこのTopHatとHISAT群の中ではHISAT2の使用が推奨されている。にもかかわらず、結構な割合でTopHatが使われているよねっていう。
STARはこれには含まれていないが、現在は結構広く用いられているようで、HISAT2かSTARか、といった具合であるように見える。
最近ではマッピングせずにそのリードのまま発現変動解析を行ったりすることも流行っているので必ずしもマッピングが必須というわけではない。ただマッピングが不要になるかと言えば、そうはならないだろう。
おそらく読者はこのSTARとHISAT2、どちらを使ったほうがいいのか、迷うことがあってこの記事にたどり着いたと思うので、どちらが優れているかを簡単にまとめておく。
高性能ハードウェア要求ではHISAT2の勝利
STARは非常に大量のメモリを消費する代わりに高速にマッピングを行えるというのがウリである。実際、私もSTARで2つほどゲノムのマッピングを行ったが、64GBメモリのパソコンギリギリでなんとかインデックスの作製ができるといった具合だった(おおよそ55GB使用)。ただしマッピングは高速で、(あくまでも参考だが)4Gbの結果をマッピングするのに10分程度しかかからなかった。
HISAT2もHISAT2でかなり高速な部類のソフトウエアである。時間としては確かそうかわりはなかったはずである。ただしこちらは省メモリ。STARほどメモリを必要としないため、そこらのデスクトップPCでもやろうと思えば十分にできるであろう。
こういった、要求されるPCスペックなどを鑑みるとHISAT2のほうに軍配が上がるだろう。
RNA-seq解析をするDRY解析のラボでは十分なハードが与えられているはずである。64GB以上あるようならば十分なので、STARを試してみても良いだろう。ただ後述のようにソフトウエア自体の精度・性能面において両者に違いが出てくるためきっちり考えて使う必要がある。
マッピング率ではSTARの勝利
次にマッピング率で見ていこう。幸いにもこのマッピング率をSTARとHISAT2で比較した論文が出ていたことから、そのデータから抜粋して見てみることとする。この論文ではいくつかの種類の乳がん細胞のRNA-seqデータを用いて解析している(Raplee et al., 2019) 。
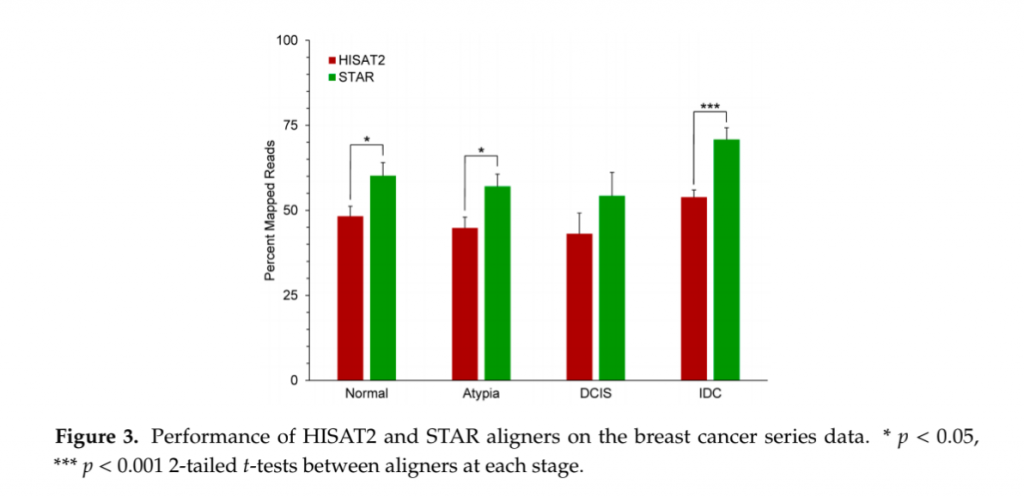
(Raplee et al., 2019 よりCC-BY 4.0に基づき引用 改変なし)
結果ははっきりしていてSTARのほうが有意にマッピング率が高い。もしHISAT2を使っていてマッピング率が低いと感じるようなのであれば、まあサンプルの問題やQCの問題も十分にあるかもしれないが、マッピングのソフトウエアだけでこれだけ違うのであれば、STARを試してみるのも良いかもしれない。ただこのデータだけ見ると、例えリードが変なところにはっついていたとしてもまあ”マッピング率”は高いよねっていう話になってしまう。
HISAT2では偽遺伝子にマッピングされやすい
そこで著者らは「本来”遺伝子”の場所に貼り付くべきであるリードが”偽遺伝子”にどれだけ貼り付いてしまっているか」を確かめた。
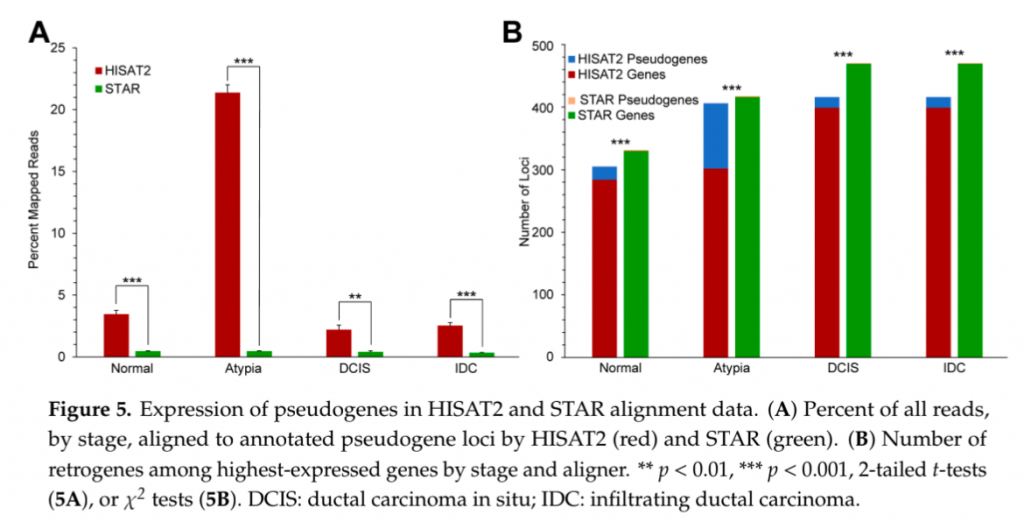
偽遺伝子の定義や選定は Raplee et al., 2019 を参照のこと。
(Raplee et al., 2019 よりCC-BY 4.0に基づき引用 改変なし)
この結果も一目瞭然で、HISAT2の偽遺伝子への誤った貼り付きがSTARよりも有意に高いことが伺える。つまるところ、HISAT2によってマッピングを行ったあと、発現変動解析などを行った場合その結果もまた大きく異なってくる可能性が存在するということである。
STARを使おう
……といったようなことから、私はもしRNA-seqのマッピングを何でやろうか迷っている人に対してはSTARを使うことを勧めたい。もちろんHISAT2も多く使われているため、ラボの人がHISAT2使ってるから私も……という人もいるだろうし、別に悪いことだとは思わない。
ただ、リソースがあるならSTARを使ったほうがより正確なマッピングができそうだよねって私は思う(今回説明は省略しているが、STARではエキソン-イントロン構造を持つ遺伝子に対するマッピングに強いアルゴリズムを採用している)。
STARについては大した使い方ではないが、次回備忘録として簡単に使い方とコマンドをまとめておこうと思う。
RNA-seqに関する記事
- SRA Toolkitの使い方 ~fastq-dumpでSRAファイルをダウンロード~
- PRINSEQ++(plusplus)でRNA-seqのRAWデータのQC(クォリティコントロール)をする
- RNA-seqデータをSTARでゲノムデータにマッピングする
参考文献リスト
- Simoneau, J., Dumontier, S., Gosselin, R., & Scott, M. S. (2019). Current RNA-seq methodology reporting limits reproducibility. Briefings in Bioinformatics.
- Raplee , I. D., Evsikov, A. V., & Marín de Evsikova, C. (2019). Aligning the aligners: Comparison of rna sequencing data alignment and gene expression quantification tools for clinical breast cancer research. Journal of personalized medicine, 9(2), 18.





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)


16GBのmac miniしかないとか、なんてひどいラボなんだ。
ありえないぜ。
r.k. さん
無限に時間がある場合はmac miniであっても解析は不可能ではありませんが……
有限な時間で手早く研究を進めるためにもぜひRNA-seq解析は自前のワークステーションでやることをおすすめします。
スパコンを必要とするまでもない解析作業であるため。
ピンバック:RNA-seqデータをSTARでゲノムデータにマッピングする – Kim Biology & Informatics