
タイトルの通り。これからハイギョのRNA-seq解析などをしたいという稀有な研究者に向けたメモ。
BLAST
正直BLASTするだけならそこまでスペックはいらない。他のゲノムと比べると時間がかかるが、数コアでも十分可能。
STAR を用いた RNA-seq マッピング
RNA-seqでもTrinityなどでdenovo assemblyをしたいというような人であれば例えば32スレッド/64GB程度のパソコンでも十分に可能。
ただ、STARのようにメモリバカ食いソフトを使う場合、128GB RAMを積んでいたとしても足りない。
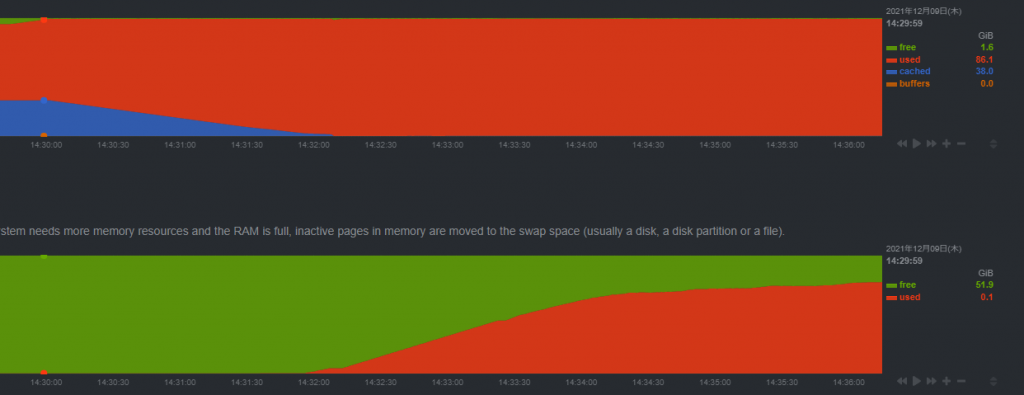
当然だがSWAP領域を使うとSSDが痛むし、IO Waitがかかって処理速度は大幅に落ちる
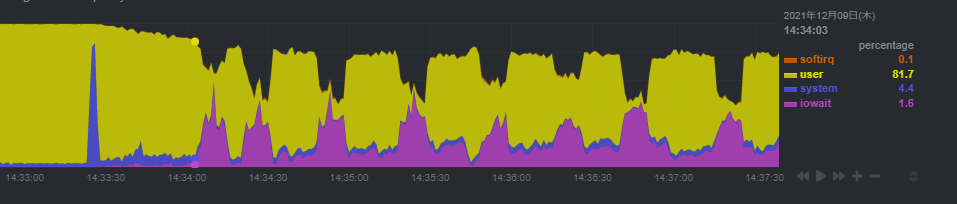
結局スワップ領域を200GB追加してなんとか9時間(32スレッド、M.2 NVMe SSD)で一番時間を食うフェーズの計算を終えた。
が、
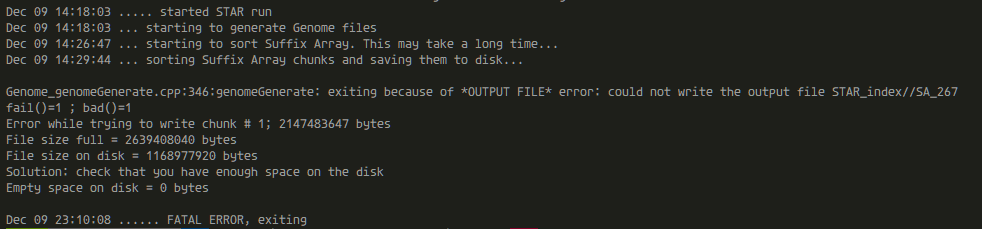
ラボに朝来て崩れ落ちた
2.6TBの保存領域が必要だけど全然足りないよというエラー。マジか。2.6TBもIndexに必要なのか……2TB SSDを使っているのでどう頑張っても足りない。厳しい。
これだけSSDを痛めて計算させたのに……またやり直し……
ということで4TB SSDを購入することに。年末まで予算少し残しておいてよかった。
で、4TBのSSD、8TBのHDDまで増設して改めて確かめるも、
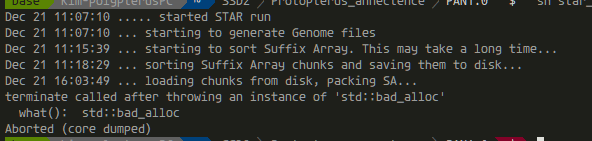
えええ……死んでる……
RAMを食い尽くすようなことはなかったと思うんだがと思いログを遡るも、
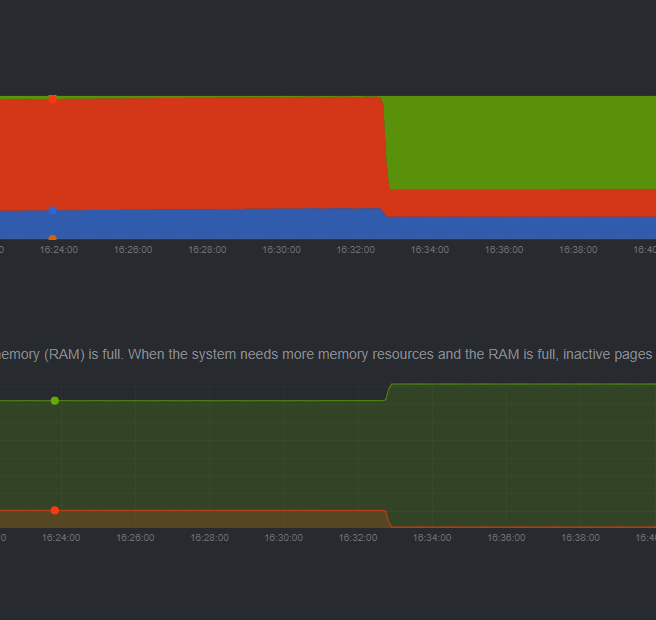
結論: STARでハイギョのRNA-seqマッピングは無理。
Hisat2 を用いた RNA-seqマッピング
もうひとつ主流のマッピングソフト(アライナー)にHisat2がある。以前少しSTARと対比して紹介した。
最近また新しいバージョンが出た(2.2.1)。今回はそれを用いてインデックスの作成を行った。こちらはマッピングの正確性(精度)では若干劣るが、ハードウエア要件が低い。
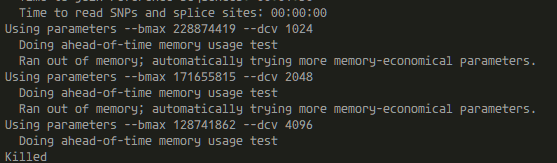
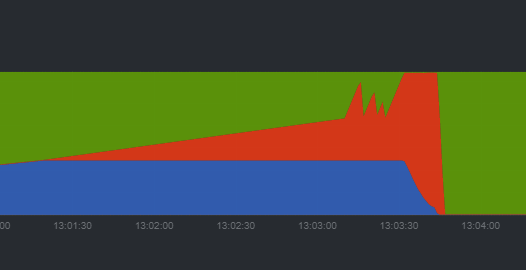
SSDに同様にSWAP領域を確保し、インデックス作成。最初にスワップ領域まで使用したが(128GB<)、その後はRAMのみで処理しきれているようだ。
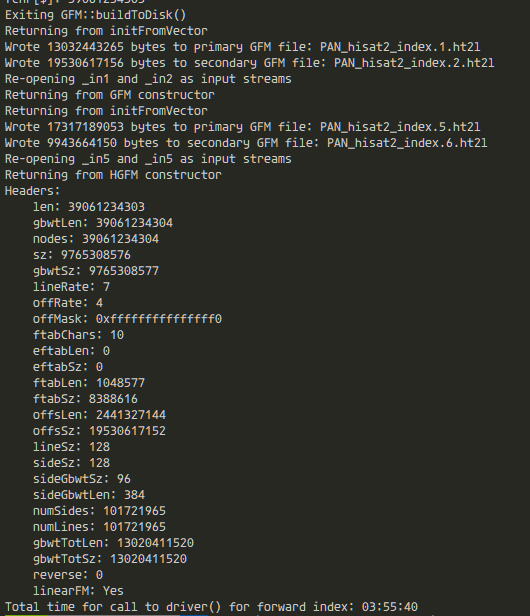
その後マッピングも問題なくできたが、samtoolsでBAMへの変換はできるものの、Indexの作成はできなかった。
また、FeatureCounts(Subread)についてもGTFの読み込み段階でSegmentation fault が生じた。
ううむ……いつも使ってるパイプラインが使えない。
ただこちらもStringtieを使うことで問題なくカウントは可能であった。
色々と問題はあるソフトは存在するが、動作するソフトもあるため、それらをうまく使う必要がでてきそうだ。
結論
128GB RAMがあれば最低限Hisat2でRNA-seqのマッピングは可能
多数のコアがあれば尚良




![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)


