
FeatureCountsを使ってマッピングしたRNA-seqリードをカウントしようとした時、どうしても全てが「0」カウントで出力されることがあった。
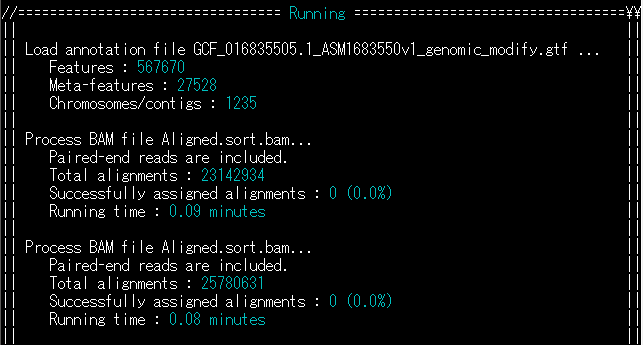
BAMファイルを調べる。
$ samtools view Align.bam | lessすると、GFFファイルとはぜんぜん異なる染色体の番号が……よくよく元のSTARのパラメータを記したshファイルを読み返すと、少し前のバージョンのゲノムを指定していた。そのせいでファイル間で齟齬が生じていたのだ。
つまり、このようなエラー(Successfully assigned alignments : 0 (0.0%))が出る場合、まず染色体番号などを見返す必要がある。
結構ググるとそういった問題が報告されていた。





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)

