
はじめに(SRAとは)
これから何回かに分けて自分で学びながら忘備録としてSRA Toolkitの使い方を残しておこうと思う。
SRA(Sequence Read Archive)を解析する方法として、SRA Toolkitを使う方法がある。というか、使わざるを得ない状況になってきている。
2022年現在、NCBIのデータベースは登録されたRNAシーケンスデータなどのFASTQファイルを直接ブラウザからダウンロードすることができない。というのも非常にそのファイルが大型であるため、ストレージを圧迫してしまうためだ。(ただしDRA,ERAといった別のデータベースでは可)
そこでSRA形式というものが開発された。それは約1/10にファイル容量を抑えることができる。現在それを用いることが必須となってきている。逆に言えばこれを使うといちいち大容量となるFASTQのファイルを全て落としてきて解析する必要がなくなる場合がある(部分的な配列を取得する場合など)。全ての配列が欲しい場合もこのSRA toolkitを使うことでダウンロードすることができる。
SRA Toolkitのダウンロードとインストール
SRA Toolkitはここからダウンロードできる。基本的に本記事ではUbuntu(Linux)での使い方を説明する。macについてもUNIXなのでほぼ一緒のはず。Windowsはそもそも解析に向いていない。
.tar.gz で落とされるので解凍。
PATHを通す。通さない場合は、絶対パスもしくは相対パスを使う必要がある。
PATHの通し方がわからない人が多いので、bashを使っている場合の例を記す。
$ nano ~/.bashrc # もしくは.bash_profile, profileなどなど。自身の環境に合わせるで、エディタが起動するので、
PATH="$PATH:/path/to/dir/"を末尾に追加して保存。当然だがpath/to/dirをそのまま打ってもうまくいかないので、適宜自分がダウンロードしたSRA-toolkitのディレクトリの絶対パスを指定してやる。
PATHを通さない場合、例えば、fastq-dumpを使う場合、
$ ./fastq-dump
$ ~/path/to/dir/sra-toolkit/fastq-dumpのようにする。
Windowsユーザーは普通にexeの模様。
SRA Toolkitの設定
ダウンロードされるファイルはデフォルトで以下の場所に送られるようだ。
Linux: /home/[user_name]/ncbi/public
Mac OS X: /Users/[user_name]/ncbi/public
Windows: C:\Users\[user_name]\ncbi\public
SRA-toolkitの使い方
Linuxで例を示す。ダウンロードしてきて解凍したフォルダのうち、binディレクトリに入ってそこでターミナルを開く(何もないところを右クリックで「端末で開く」があるのでそれで開ける)か、普通にbinのディレクトリまでcdコマンドで進む。
そこでターミナルに
$ ./fastq-dump -X 5 -Z SRR390728と入力し、実行。
Windowsの場合は
$ fastq-dump.exe -X 5 -Z SRR390728しばらくすると、NCBIに接続し、SRR390728に接続、最初の5つ分のデータを切り出して持ってくる(-X 5の引数がこれに当たる)。さらにそれをターミナル上に映し出す(-Z 引数がこれに当たる)。
実行結果は以下の通り。
————実行結果————
$ ./fastq-dump -X 5 -Z SRR390728
Read 5 spots for SRR390728
Written 5 spots for SRR390728
@SRR390728.1 1 length=72
CATTCTTCACGTAGTTCTCGAGCCTTGGTTTTCAGCGATGGAGAATGACTTTGACAAGCTGAGAGAAGNTNC
+SRR390728.1 1 length=72
;;;;;;;;;;;;;;;;;;;;;;;;;;;9;;665142;;;;;;;;;;;;;;;;;;;;;;;;;;;;;96&&&&(
@SRR390728.2 2 length=72
AAGTAGGTCTCGTCTGTGTTTTCTACGAGCTTGTGTTCCAGCTGACCCACTCCCTGGGTGGGGGGACTGGGT
+SRR390728.2 2 length=72
;;;;;;;;;;;;;;;;;4;;;;3;393.1+4&&5&&;;;;;;;;;;;;;;;;;;;;;<9;<;;;;;464262
@SRR390728.3 3 length=72
CCAGCCTGGCCAACAGAGTGTTACCCCGTTTTTACTTATTTATTATTATTATTTTGAGACAGAGCATTGGTC
+SRR390728.3 3 length=72
-;;;8;;;;;;;,*;;';-4,44;,:&,1,4'./&19;;;;;;669;;99;;;;;-;3;2;0;+;7442&2/
@SRR390728.4 4 length=72
ATAAAATCAGGGGTGTTGGAGATGGGATGCCTATTTCTGCACACCTTGGCCTCCCAAATTGCTGGGATTACA
+SRR390728.4 4 length=72
1;;;;;;,;;4;3;38;8%&,,;)*;1;;,)/%4+,;1;;);;;;;;;4;(;1;;;;24;;;;41-444//0
@SRR390728.5 5 length=72
TTAAGAAATTTTTGCTCAAACCATGCCCTAAAGGGTTCTGTAATAAATAGGGCTGGGAAAACTGGCAAGCCA
+SRR390728.5 5 length=72
;;;;;;;;;;;;;;;;;;;;;;;;;;;;;9445552;;;;;;;;;;;;;;;;;;;;;;;;;;;;;;446662Configの使い方
Configをいじることで先程のダウンロードのファイルの場所(作業場所)を変えることができる。
ノートパソコンでやっていて、ストレージ容量が大きくないために外部ストレージなどに作業場所(ワークスペース)を変えるときなどにConfigをいじる。
方法は先程のbinディレクトリ下において、
$ ./vdb-config -iと実行。
以下のような画面となる。
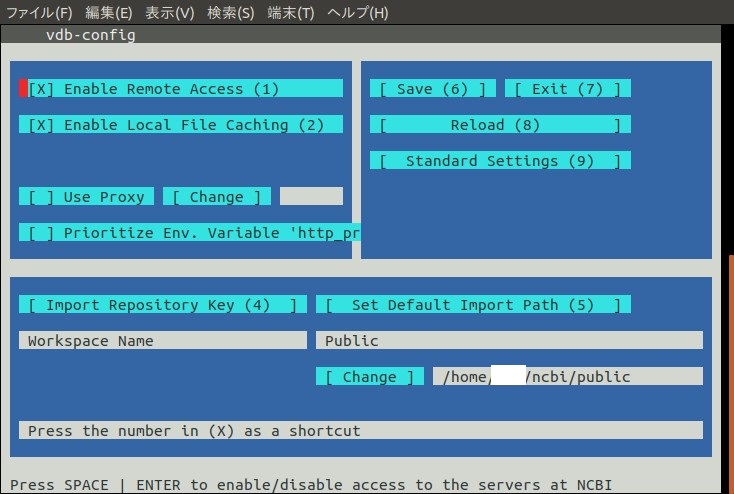
赤い四角がポインタで現在どこを指しているかだ。それをTabキー、もしくは矢印キーで移動させていく。
ワークスペースを変える場合は[ Change ]までポインタを進めてからEnter。
以下のような画面となる。
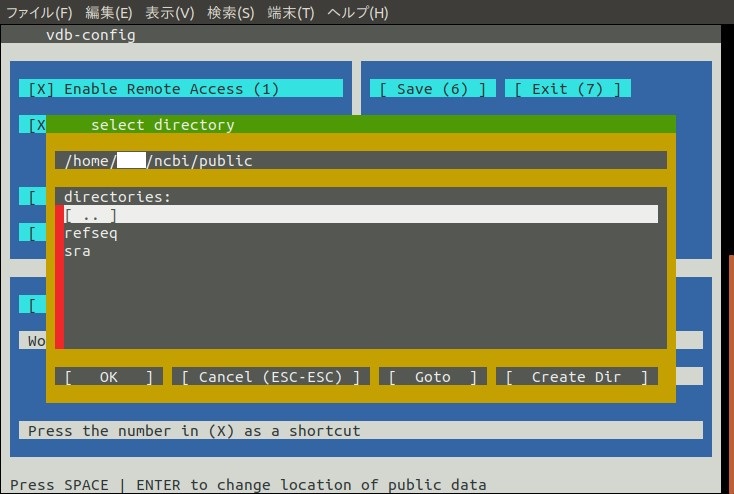
上の階層に行く場合は[ . . ]を選択する。
変更したら、SaveしてExit。これでワークスペースを変えることができる。
次は実際に配列解析を行ってみる
SRA Toolkitの使い方 ~fastq-dumpでSRAファイルをダウンロード~
参考文献
- 坊農秀雅(2017), “Dr. Bono の生命科学データ解析”, p107-108
- https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc&f=std





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)

