
RNA-seqのマッピングにはいろいろなものが開発されている。中でも有名なものがBowtie系、HISAT2、そしてSTARである。
現在主流なのはHISAT2とSTARであり、性能としてはSTARのほうが上であるが要求マシンスペックはSTARのほうが高い。
参考: RNA-seqのマッピングツールはSTARかHISAT2か
STARの論文によると、他のマッピングツールよりも偽陽性が低く、高速であるそうな。
昔RNA-seqを使っていた頃はHISAT2を使っていたが、RNA-seqのためにパソコンを用意し、メモリも64GBあればなんとかなるでしょと思い、STARを使っている。5Gb(ギガベース)x2程度のRNA-seqのペアエンドRAWデータをマッピングするのに10分もかからない。(そこはリファレンスゲノムの大きさにも依存するとは思うが)
今回はそのSTARのインストールの仕方、使い方を軽くまとめておく。いつものことながら、Ubuntu 19.10 , Ubuntu 20.04 LTS を対象に話をすすめる。
STARのインストール
UbuntuにおけるSTARのインストールは非常に簡単である。というのも、パッケージが配布されているからだ。
$ sudo apt update $ sudo apt upgrade
で一応最新にしておく。
$ sudo apt install rna-star
これでインストールが完了だ。面倒なことは一切ない。ただ遺伝研スパコンなどの管理者権限を持たない環境では別の方法でインストールする必要があるので注意。
STARの使い方(アノテーション情報なし)
インデックスの作成
HISAT2にしろ、まずリファレンスゲノムに対してインデックスを作成する必要がある。STARもこのインデックスの作成が一番時間がかかり、リソースも消費する。
詳しい方法としてはマニュアルが存在するのでそちらを参照するとして、手軽に始めたい人間に対して私が使っているコマンドを書き留めておく。
例として以下のようなコマンドによってインデックスを作成することができる。これは32スレッド、64GB RAMがあるマシンの例である。
$ STAR --runMode genomeGenerate --runThreadN 32 --genomeDir index_Reference --genomeFastaFiles Reference.fa --limitGenomeGenerateRAM 55000000000
一つずつコマンドを見ていこう。
- –runMode: これに genomeGenerate を与えることでインデックスの作成をすることができる
- –runThreadN: 使用するスレッド数。マルチスレッド対応なので多ければ多いほど早く作業が終わる
- –genomeDir: 出力ファイルが保管されるディレクトリの名前。後々の実行で使うので、必要十分かつ他のインデックスと重複しない名前をつけること。先にディレクトリを作成しておく必要がある。
- –genomeFastaFiles: リファレンスゲノムのFASTAファイルの指定を行う
- –limitGenomeGenerateRAM: このインデックスの作成に割り当てるRAM(メモリ)の量を指定する。できるだけ大きいほうが良い。ちなみにPCのスペックよりも大きいメモリを指定するとエラーを吐くので注意。
こんな感じで放置しておけばインデックスを作成できる。
今回はGTFファイルを入力に使わない方法を紹介している。GTF/GFFファイルを与えてやればマッピングの際に遺伝子ごとのカウントも自動でやってくれる。今度やる機会があったら追記を行う。
GTFファイルを使ってインデックス作成
–2020/04/30 追記–
マニュアルいわく、2.4.1a以降ではマッピングの段階でGTFファイルを読み込ませる手段があるらしい。ちなみにaptを使ってインストールすると2020/04/30の段階では最新バージョンである2.7.3aがインストールされる。
インデックス作成段階で、
--sjdbGTFfile /path/to/annotations.gtf
のオプションをつけるだけでGTFを読み込ませることができる。読み込ませることによってスプライスジャンクション(イントロン/エキソンの切れ目)を考慮することができ、マッピングの精度が大幅に上昇する。
追加分も含めると以下のコマンドとなる。適宜値は自分の環境に合わせて変更すること。
$ STAR --runMode genomeGenerate --runThreadN 24 --genomeDir index_Reference --genomeFastaFiles Reference.fa --limitGenomeGenerateRAM 55000000000 --sjdbGTFfile annotations.gtf
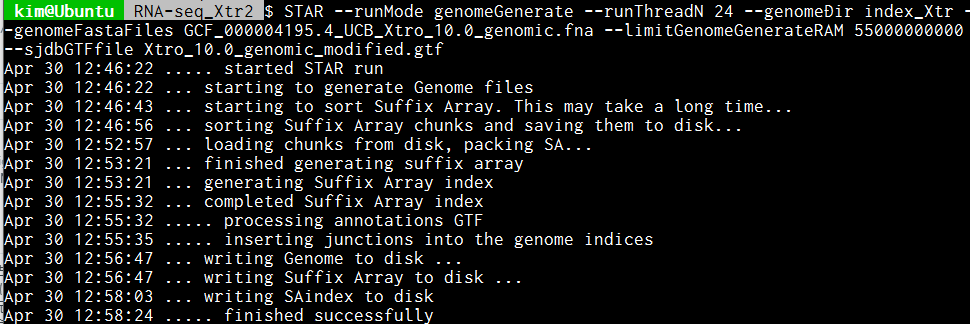
参考までに、ネッタイツメガエル(Xenopus tropicalis)のゲノムデータ(total length (Mb): 1451.3)のインデックスをGTFファイル付きで作成したところ、Ryzen9 3900X(12コア24スレッド)、64GB RAM(使用に指定したのは55GB)の環境下ではおよそ12分かかった。
(2021/05/25追記: ポリプテルス(Polypterus senegalus)のゲノムデータ(median total length (Mb): 2443.28)も同様に作成したところ、Ryzen9 3950X(16コア32スレッド)以下同様でおよそ17分かかった。)
マルチスレッド対応なのでボトルネックはCPUである。まあ一度インデックスを作ってしまえばその後作り直すことはそうそうないのでここで時間がかかっても特別問題にはならないだろう。
GFFからGTFへの変換は以下のコマンドでできる。(参考: gff3 ↔gtf の変換 )
$ gffread annotation.gff -T -o annotation.gtf
マッピング
無事にインデックスの作成が完了したら、次は実際にRNA-seqデータをマッピングする。マッピングの際に注意すべき点として、予めクオリティコントロールを行っておく必要があるということ。あまりにも短い配列や信頼度の低い配列が混じっていると不正確なマッピングや余計に時間がかかったりする可能性があるため、予め取り除いておく。
参考: PRINSEQ++(plusplus)でRNA-seqのRAWデータのQC(クォリティコントロール)をする
以下のコマンドで実行可能である。
$ STAR --runThreadN 8 --genomeDir index_Reference/ --readFilesIn ./trim_sample_good_out_R1.fastq ./trim_sample_good_out_R2.fastq
念の為解説しておくと、
- –genomeDir: 先程作成したIndexが入ったディレクトリ
- –readFilesIn: ペアエンドのFASTAファイルを2つ、スペース区切りで入力。PRINSEQ++でQC後ならばgoodがついているもの
だけでOKである。
案外すぐに終わり、実行ディレクトリ以下にログファイルなどが生成される。
例えば、Log.final.outでは以下のようなマッピングの細かい情報が得られる。あまりにもマッピング率が低いようならば見直して見る必要があるかもしれない。
Started job on | Nov 18 21:51:08
Started mapping on | Nov 18 21:52:39
Finished on | Nov 18 22:01:44
Mapping speed, Million of reads per hour | 162.25
Number of input reads | 24562503
Average input read length | 191
UNIQUE READS:
Uniquely mapped reads number | 21964270
Uniquely mapped reads % | 89.42%
Average mapped length | 189.84
その他SAMファイルが出てくると思うが、それを今後リードカウントしたりするのに使う。


![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)




ピンバック:RNA-seqのマッピングツールはSTARかHISAT2か – Kim Biology & Informatics