
先日またRNA-seqを外注した。
FASTQCでクォリティチェックを行ったところ、以下のように、”Overrepresented sequences”に関する表示が出ていた。なにやら”GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG”のリードが2万近くあるとのこと……
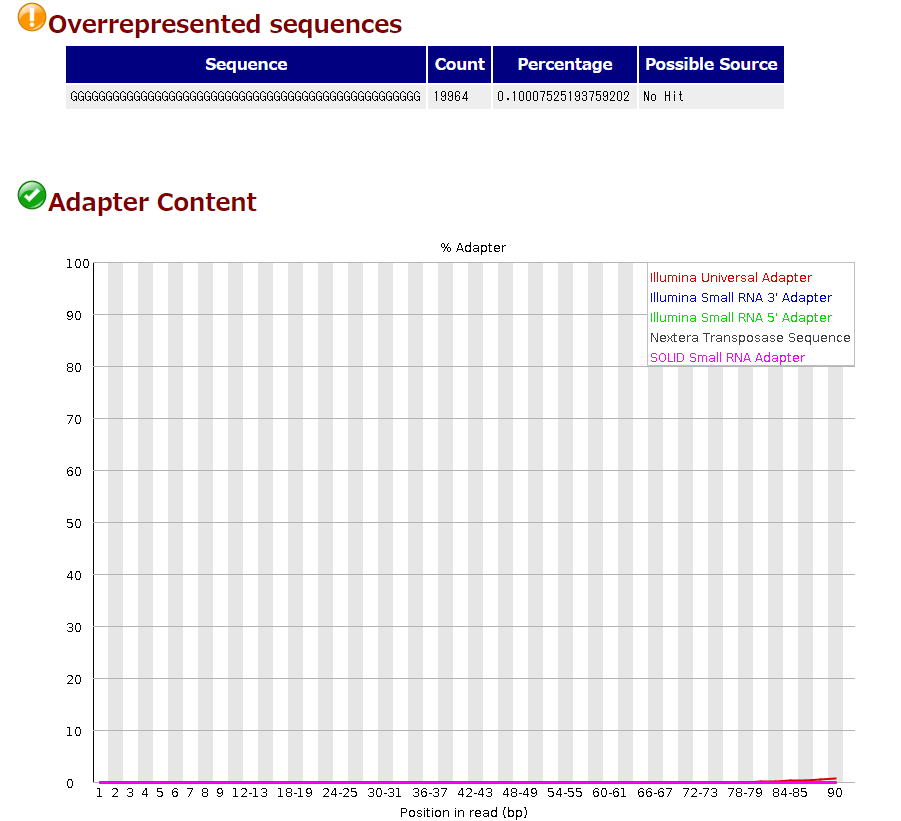
あとビミョーにアダプター配列も検出された。
もちろん、GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGなんてBLASTかけたところで出てこなかったし、これが何なのかわからない。おそらくライブラリかシーケンスのエラーだろうと思って調べてみると、Illuminaのシーケンサ(NovaseqやNextSeq)特有のエラーらしい。読み取りがうまく行かなかったリードがこのGGG……になるというわけだ。
参考: Illumina 2 colour chemistry can overcall high confidence G bases
fastpでポリG配列を取り除く
こんなものを残した状態でマッピングするのはよろしくないので、トリミングする。
参考: Trimming reads and removing adapter sequences and polyG tails
fastpを使えば、Optionを加えてやるだけで簡単に取り除ける。
$ fastp -i "$FILE"_1.fastq.gz -I "$FILE"_2.fastq.gz -o trimmed_"$FILE"_1.fastq.gz -O trimmed_"$FILE"_2.fastq.gz -g -h report_"$FILE".html -q 20 -w 16これのように、-gを加えてやるだけでポリG配列を排除できる。
その他のオプションは例えば-q 20は少なくとも20Phredクォリティスコアが必要、-w 16は使用スレッド数(最高16)、等。アダプター配列は特に指定しなくても取り除いてくれる。
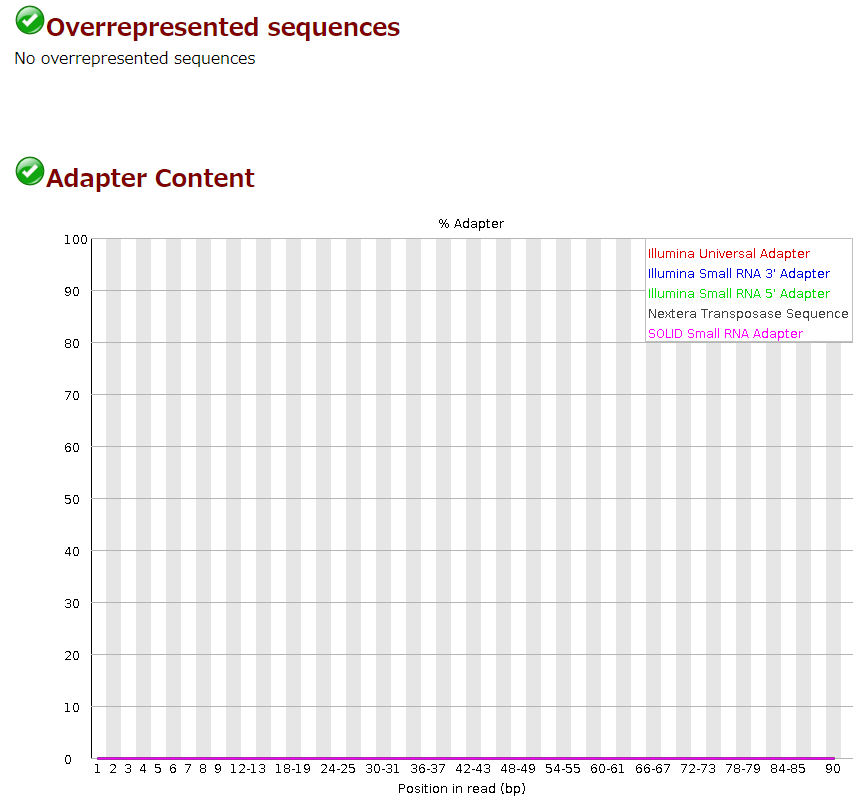
きれいに取り除けた。
いやしかし、今回の発注したデータはポリGが多かったしその分エラーが多かったということ……なんかもったいないなあ。




![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)


