
参考: Sequence length from Fasta
マルチFASTA で、それぞれの配列の長さを出してほしい時がある。
そういったときに簡単に出力してくれるのがbioawkだ。
conda install bioawkで、仮想環境上にインストール。
bioawk -c fastx '{ print $name, length($seq) }' < input.faで、出力してくれる。
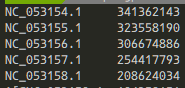
全部出すと長い場合、パイプで繋いでやって欲しい部分に絞ってやれば良い。
bioawk -c fastx '{ print $name, length($seq) }' < input.fa | head -n 20<br>$ bioawk -c fastx '{ print $name, length($seq) }' < input.fa | tail -n 20




![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)

