
今回は複数種における複数の遺伝子の塩基配列をアラインメントし,最尤法を用いた系統樹推定を行いたい.
タイトルにもある通り,今回はUbuntuを扱って解析を行う.mac等は他にTOGOTVなど色々解説しているところがあるので.
環境
環境は以下の通り
- Ryzen5 3400G (Ryzen9 3900Xでも動作確認)
- メモリ 64GB
- Ubuntu 19.04 (Ubuntu 20.04 でも動作確認)
MAFFTでアラインメント
MAFFTのインストール
MAFFTとはマルチプルアライメントのツールである.
阪大の加藤 和貴 先生が作成したプログラムで,こちらのサイトからダウンロードできる.
さて,現時点(2019/09/25)におけるMAFFTの最新バージョンは” The latest version is 7.429 (Windows), 7.427 (others), 2019/Jun. “とある.
もちろん最新のソースコードからコンパイルしてやってもいいのだが,いささか面倒くさい.左のメニューバーから「Linux」を選択してやればdevのパッケージファイルが手に入るのでそちらをダウンロードしてインストールしてやってもいいだろう.
だが,それも面倒くさい人間は
sudo apt install mafftでいける.これは以下の通りバージョンは最新版ではない.しかしバグのある7.380–7.392 ではないため,まあ問題はないだろう. sudo apt を使ってインストールすることは手軽だが,バージョンが古かったりするので注意が必要である.
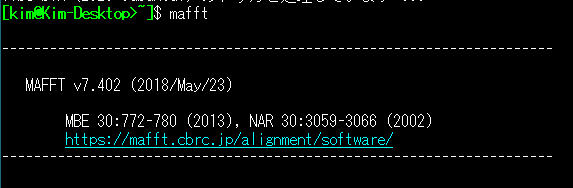
本記事ではMAFFT v7.402を使うこととする.
Online MAFFT
……と先月の執筆時点ではそうしていたのだが,作者のサイトの方でオンラインでMAFFTが使えるということでそちらを使うことにした.ちなみにローカルでアラインメントをやりたいという場合もこちらのサイトで実行するとコマンドが表示されるのでそれをメモして環境に合わせて実行することができる.
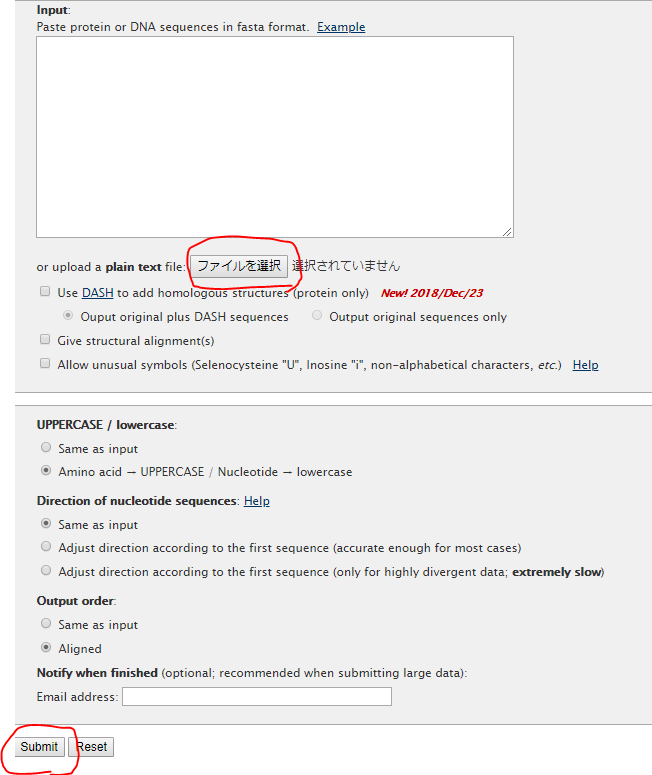
MULTI FASTAファイルをアップロードしてsubmitしてやるだけで解析をしてくれる.終わったファイルはタブの”FASTA”から表示してダウンロードしておく(コピペ).
RAxMLで最尤法系統樹推定を行う
RAxMLのダウンロード
Githubからcloneすればよい.某I先生のサイトではやりづらいから今後変わるだろうとあるが,2019年現在,Githubが主流となってきているので逆にこれからこういう配布形式は増えていくと見て良い.
前提としてGitが入っているものとするが,もし入っていない場合は
sudo apt install gitでgitをインストールする.そして保存するディレクトリに移動したあと,以下のコマンドを入力する.
git clone https://github.com/stamatak/standard-RAxML.gitこれでGitHubからリポジトリがクローンされたはずである.
RAxMLのインストール
さて,ダウンロードが終わったら次はインストールしていく.インストール方法は至って簡単で,クローンしたフォルダに移動してから
make -f Makefile.AVX.PTHREADS.gcc
rm *.oで完了である.色々あるが,おすすめはこのPTHREADSがついているもので,こちらはマルチスレッド対応となるのである程度コア数・スレッド数があるPCではこちらを優先的にインストールすべきである.
あとはパスを通すために,
nano ~/.profileで末尾に
PATH="$PATH:/path/to/dir/"と追加する(当然だが/path/to/dir の部分は自分の環境に合わせて設定すること.GitHubからダウンロードしてきたフォルダのパスを書けば良い).
書き終えて保存したら再起動するか
source ~/.profileで変更した”.profile”を読み込む.これでPATHが通った.
RAxMLで系統樹推定を行う
さて,RAxMLで系統樹推定を行っていくが,まずはマニュアルを読もう.大抵のことはここに書いてある.だがマニュアルが読めない・面倒くさい・よくわからないからこのページにたどり着いたということも重々承知である.
サクッとMAFFTでアラインメントしたファイルを解析する方法を述べておこう.
まず大前提として大事なことに「FASTAファイルの”>”の行に”(“や”[“,”,”などの文字が含まれていないこと」が重要である.これが含まれているとうまくいかない.スペースがあってもうまく認識されないので必ず”_”でスペースは埋めること.これをまず確認してほしい.
おそらく進化生物学屋さんが行うことが主だと思うが,現在の主流としては分岐部分に信頼度(confidence)であるブートストラップ値(bootstrap value)を載せることが多い.載せないこともあるが,有意差等々結局何らかの信頼性を掲載することが論文を読んでいると多い.そのため今回はBootstrapを掲載するまで行うこととし,Bootstrapを100として計算する例(-# 100)を載せる.
また,今回はアミノ酸の例としてモデルを”-m PROTGAMMAAUTO”で設定する.アミノ酸のモデルとしては定番のようなものがないらしく,オートで決めてしまえと思ってやっている.
ちなみに塩基配列の場合は様々なモデルが用意されており,それぞれどれが良いかを決める必要がある.MEGAなどでどのモデルが適しているかを判断してくれるプログラムがあるのでそれでやってみるといい(RAxMLにはないモデルが選ばれることもあるが).
RAxMLでは使用するCPUのスレッド数を指定することができるので,”-T”オプションで指定してやる.指定するスレッド数をそのPCのスレッド数以上の数を割り当ててしまうとどうやらスレッドの取り合いが起きるらしく結果として遅くなってしまうので必ず最大値はそのCPUのスレッド数に抑えておくこと.Ryzen5 3400Gは4コア8スレッドなので”-T 8″とした.(Ryzen9 3900X だったら24で最高性能が出る)
さて,これらのオプションを含めたコマンドは以下の通りである.
raxmlHPC-PTHREADS-AVX -f a -m PROTGAMMAAUTO -s MAFFT_alignment_MULTI.fasta -p 12345 -x 12345 -n HOGE -T 8 -# 100“-s”のあとはマルチFASTAファイルのパスが来る.普通にディレクトリを作ってそこにFASTAファイルを移動させてから実行させたほうが良いだろう.-p, -x の値はなんか初期値に関連しているらしく,おまじないのように12345としているものが多い.マニュアルですらも.
“-n”は出力ファイルの拡張子になるが,多分実行の識別のために使っていると思われる.途中でエラーを吐いて停止した際にHOGEの拡張子のファイルが残っていると再実行がうまくいかないのでそれを削除してやり直さなければならない.
“-f a”は高速ブートストラップおよびML解析を行うオプションである.これでいい. 解析開始である.
さて,しばらくすると
Overall execution time for full ML analysis: 13193.461918 secs or 3.664851 hours or 0.152702 daysのように表示され,解析が終わる.123もの解析を行ったので結構時間がかかったが,ML解析としては高速な部類に入る.Ryzen9買ってくれればもっと早くできるのにな~~~~~~~~~~~~~(チラッ
RAxMLでブートストラップ値を系統樹に載せる
ここだけはかなり苦戦したので英語版も載せておく.
→ English version How to make a phylogenetic tree with Bootstrap values in RAxML? coming soon!
まずは得られたファイルを確認しよう.”RAxML_bestTree.HOGE”と”RAxML_bootstrap.HOGE”が少なくともあるはずだ.これのHOGEを”tre”に変えてやってFigTree(後述)などで見ると普通に見れるが,肝心のBootstrap値が見れない.
そこで系統樹にBootstrap値を載せる必要がある.以下のコマンドでそれが可能だ.
raxmlHPC-PTHREADS-AVX -f b -m PROTGAMMAAUTO -t RAxML_bestTree.HOGE -z RAxML_bootstrap.HOGE -n BOOTSTRAPこれにより,同じディレクトリに”.BOOTSTRAP”の拡張子がついたファイルが3つ生成されるはずだ.これのうち,“RAxML_bipartitions.BOOTSTRAP”が目的のファイルである.
さて,下準備は終わったので最後に系統樹を見る・作るソフトウエアを見よう.
FigTreeで系統樹を見る
FigTreeのインストール
さて,ここからはGUIが使えることが前提となる.動作確認はWindows10でやっている(UbuntuのGUIでもできるはずであるが忙しくて確認できていない).
コンパイル済みのファイルがGitHubにて配布されているのでそちらからダウンロードする.今回は現時点で最新の FigTree v1.4.4 を使用する.
FigTreeの使い方
ダウンロードし,ファイルを展開したらメモ帳みたいなアイコンの実行ファイルがあるはずだ.
これを実行し,先程得られた “RAxML_bipartitions.BOOTSTRAP” の拡張子を”.tre”に変えた上で読み込ませる.
するとこのようなダイアログが出てくる.
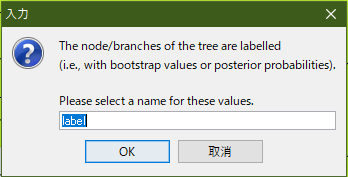
なんかノードや枝がラベリングされているからその名前をつけろっていうんで,Bootstrapに変えた上でOKを選択.別にわかればなんでもいいんだけど.
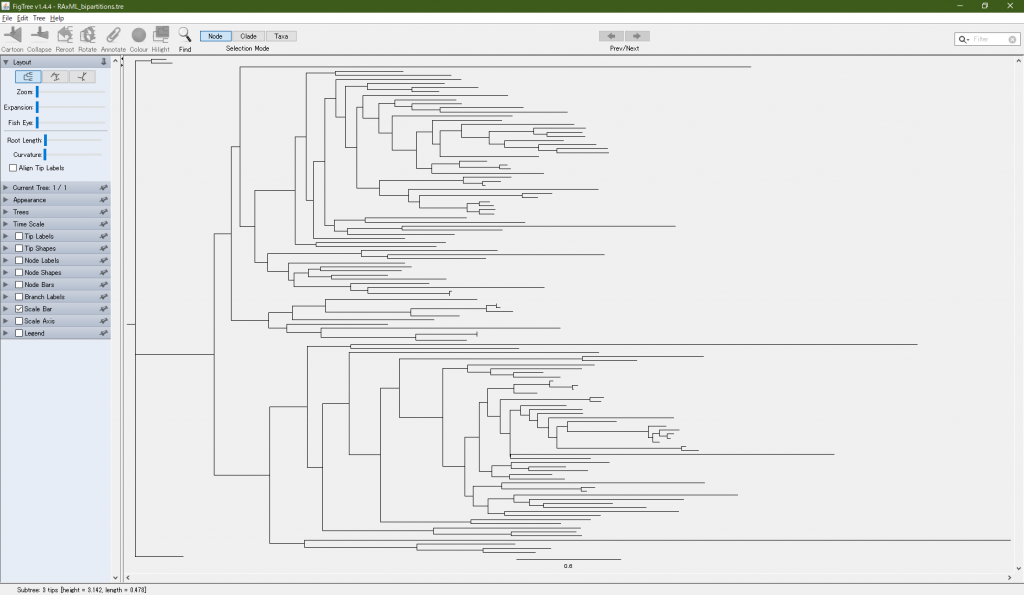
このように系統樹が表示される.今回は無根の系統樹を描きたかったので左のLayoutのところから真ん中のボタンを選択.
これで
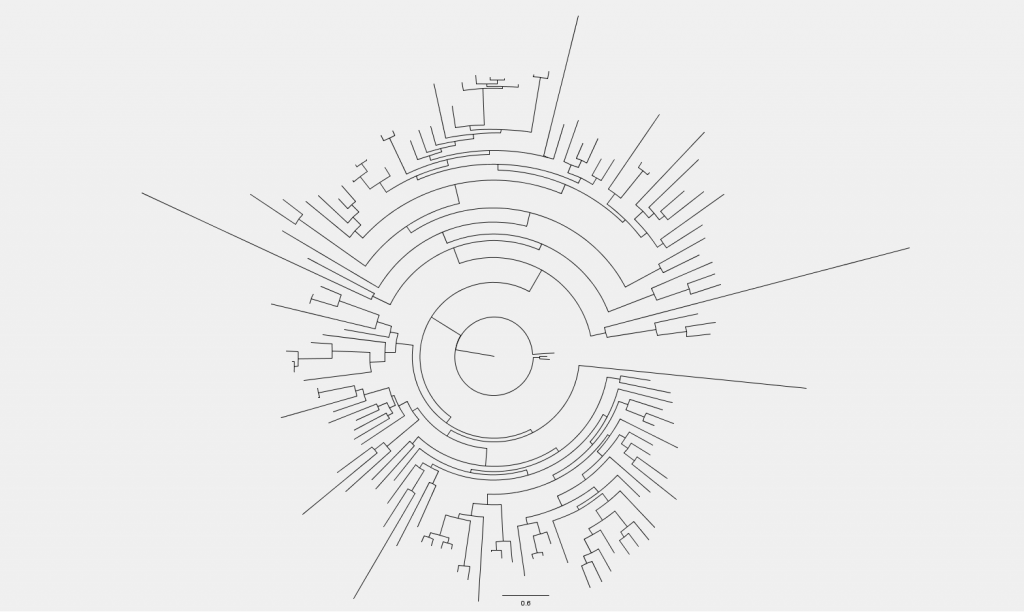
こうなる.あとは少しずつ好きなように左のツールバーから変えてやれば良い.
さて,肝心のブートストラップ値を載せるところだが,この左のツールバーにあるNode Labelsを開いてやり,その中のDisplayからBootstrapを選択する.
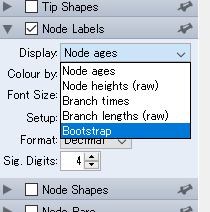
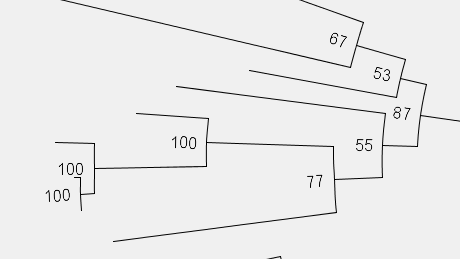
やったね.
あとはFileから各種形式に出力できる.
最後に
最後のこのBootstrap値を載せるのにクソみたいに苦労した.他のソフトではあっさりいけた記憶があるが,どうしても今回無根系統樹が描きたくてFigTreeが必要だったので苦労して試行錯誤した.
ネット上の日本語文献も結構当たったが,リンク切れがあったり情報が古かったり,浅すぎたりしたので今回この記事を書くこととした.新しく学ぶ学生諸氏はぜひ参考にしていただきたい.
わからないこと・記事に間違い等があったらコメント欄まで.





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)


ピンバック:How to make a phylogenetic tree with Bootstrap values in RAxML? - Kim Biology & Informatics
ピンバック:フリーソフトMEGAでコロナウイルスの配列解析をしよう! – 暇を持て余した情報・生物系学生へ – Kim Biology & Informatics