
導入
ヒトやマウスといったモデル生物以外のゲノムが染色体レベルで組み上がる時代に突入している。ゲノム「解読」黎明期ではゲノムを決定することで様々な生命現象が明らかになると思われていたが、実際は一次元の塩基配列だけでわかることには限界があるということがその後段々と浸透していった。そのため現在では「ゲノム解読」といったような仰々しい言葉ではなく、「ゲノム決定」という作業的な言葉を用いることが増えてきている。問題は、ゲノム配列の決定後、どのような遺伝子や領域に着目して研究するというところにある。
最近だと脊椎動物全ゲノムプロジェクト(VGP)などの国際的なコンソーシアムによるゲノム決定が盛んであり、20年前だと1種のゲノムを決定するたびにNatureの表紙を飾ったりしていたが今はもうペーパー自体が出ることも少なくなってきている。
さて、そうした「ポストゲノム時代」(これも死語になった)ではある程度自由にゲノム配列にアクセスが可能になり、大量のゲノムを比較するようなこともできるようになってきた。「あの種のゲノム配列が見たい!」「進化上でどこでこの遺伝子は獲得されたのか?」「この遺伝子に似た遺伝子はあるのか」「塩基配列の変異を見たい」といったようなことにはリファレンスゲノムがよく用いられる。
リファレンスゲノムとは、その生物種を代表した1つのゲノム配列のことである。NCBIやEnsemblなどではそのリファレンスゲノム1つに対してアノテーション(遺伝子の注釈)が行われている。これは絶対普遍というわけではなく、より精度の高いものが出たりすると入れ替わったりする。
今回は2024年に新しくなったNCBIのゲノム配列取得方法について初心者向けの解析を書いていきたい。
探したい種のゲノムを見つける
NCBI Genomeのサイトから基本的にサーチする。
具体的な属名(種名)がわかっている場合
今回は”Polypterus senegalus“のリファレンスゲノムがほしい! という例を見ていこう。ついでに他のポリプテルスのゲノムが出ていないかもチェックしたい。
「Search term」のところに種名を打ち込んでいくと、登録されているゲノムの属名から種名が出てくる。
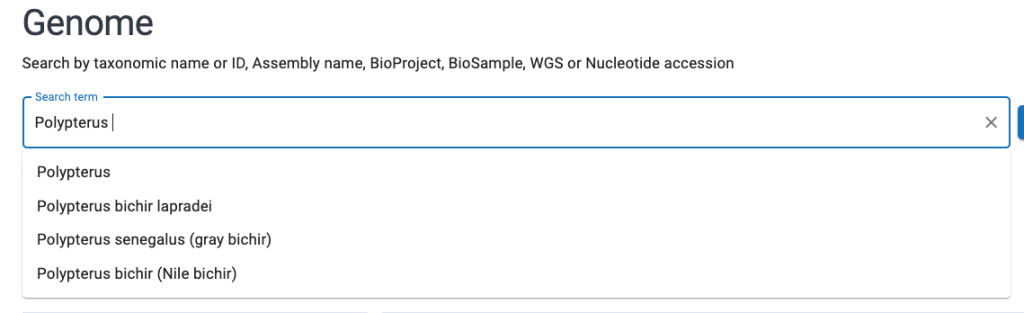
ここでは一番上のPolypterusというリストをクリックする。
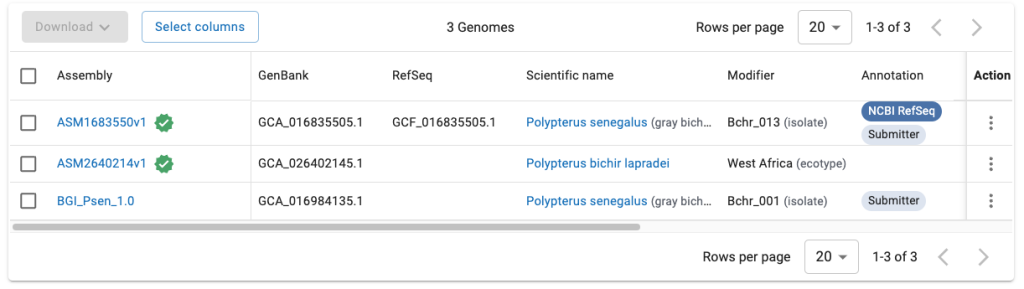
現在NCBIに登録されているポリプテルスのゲノムは3つあることがわかった。しかしScientific nameを見ると、Polypterus senegalus が2つもあることがわかる。どちらが「リファレンスゲノム」なのだろう? 一番左のカラムの緑のバッヂがついているものがリファレンスゲノムである。基本的に一種に1つしか存在しない。
では、リファレンスゲノムにアノテーションはついているだろうか? RNA-seq解析を手軽に行うにはアノテーションが必要だからだ。それは一番右のカラム、Annotation を見ると「NCBI RefSeq」と書かれているものがある。これはNCBI側のフォーマットに則って作られたアノテーションが存在することを示している。
ある系統のゲノムを網羅的に探したい場合
サーチボックスに今度は「Caudata」と入力してみよう。Caudataは両生類の有尾目のことである。すると、有尾目に属する生物種のゲノムが出てくる。
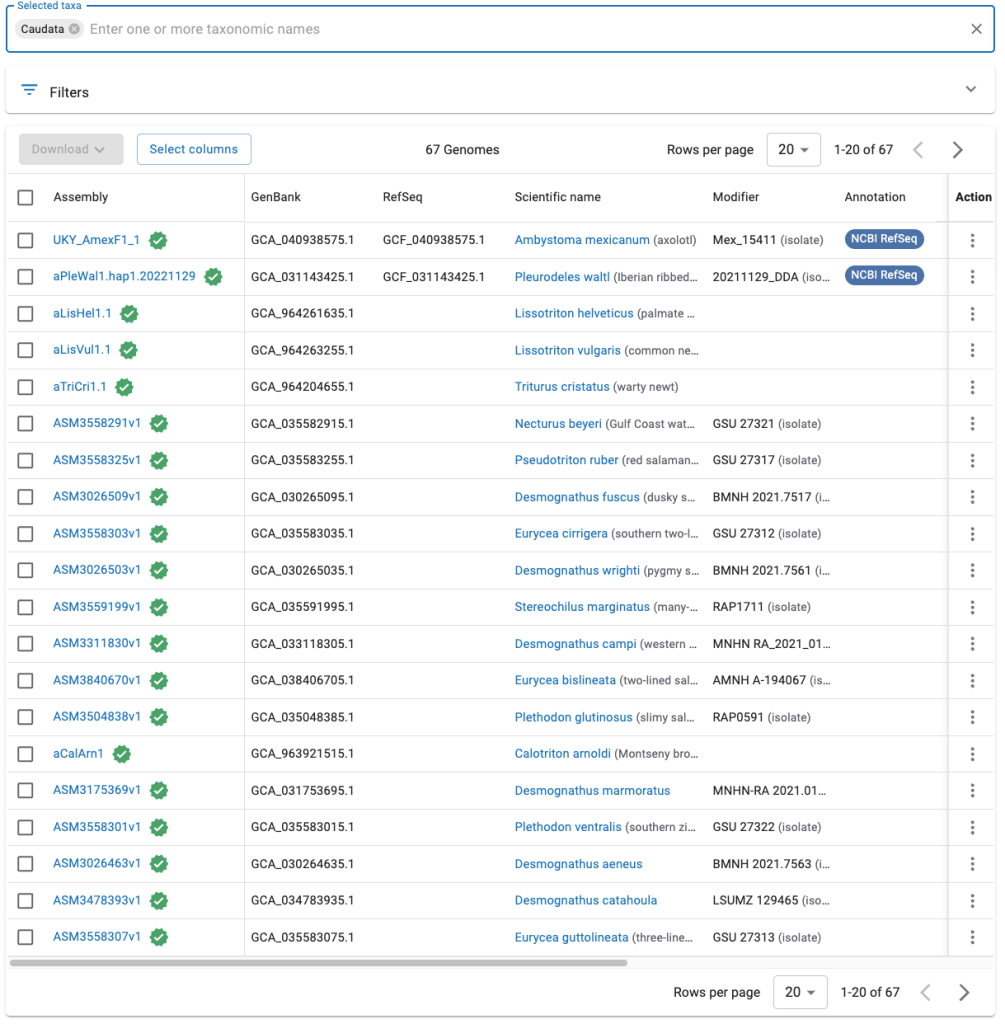
すごくたくさん登録されている! すごい! 緑のバッヂもあるからリファレンスゲノムが大量に決定されている! と糠喜びするのもいいが、このご時世、品質が悪い・不誠実な登録が大量にされている例があり、これはその最たる例である。有尾目はゲノムサイズが大きいことで知られている。Select columsをクリックして表示するものを選ぶ。今回はSize (Mb)を見る。
Applyを押すと適用される。
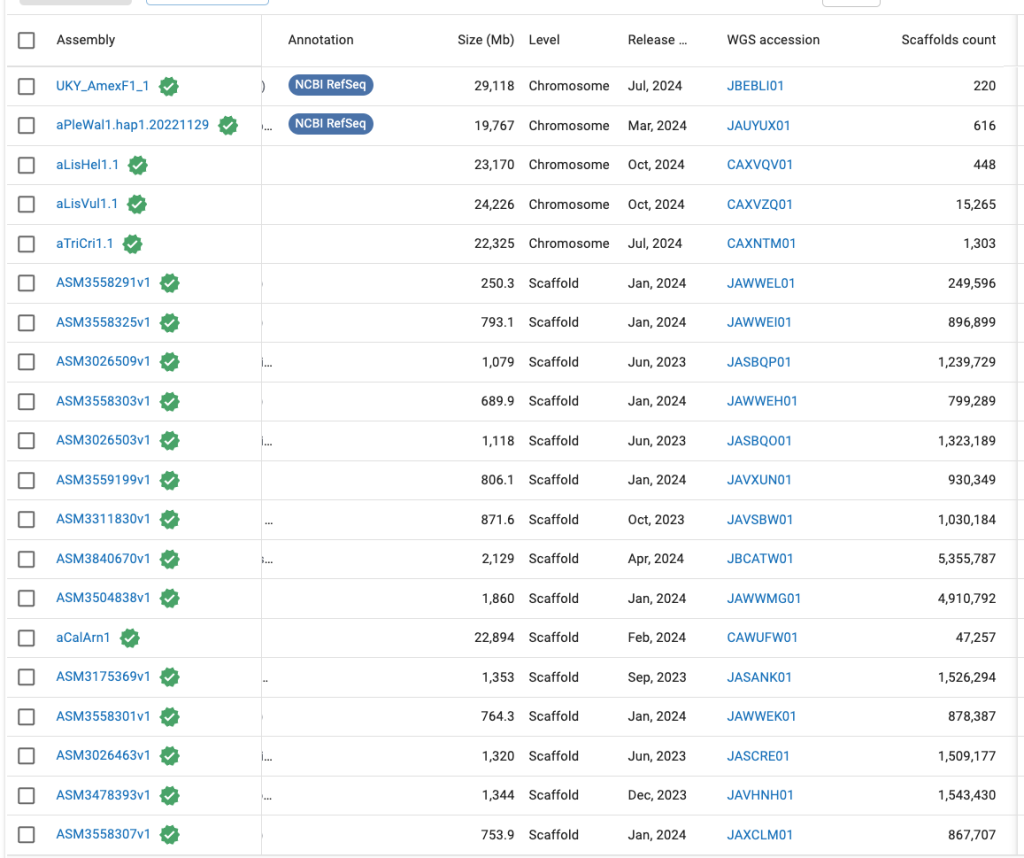
一番上がアホロートル(論文も出ており信頼できるゲノム)、二番目がイベリアトゲイモリ(信頼できる)3つ目以降も「VGPルール」に則ったアセンブリ名(両生類、Amphibiansだとaの接頭辞がつく)ことから、信頼ができると見てよいだろう。
これらのゲノムサイズを見るとどれも20 Gb 以上であり、有尾目のゲノムはそのくらい大きい傾向があることがわかる。それ以下のものを見てみると 250 Mb とかいう、あり得ない値のゲノムが大量に載っていることがわかる。つまり、ニセモノだ。こういうニセモノもNCBIにはリファレンスゲノムとして含まれていることに留意し、ゲノム情報の背景、つまり生物自身についてきちんと理解した上で扱う必要がある。機械任せになんでもかんでもしていると足元すくわれるよ。
ゲノムの情報を見る
ではそのアセンブル(ゲノム配列)にアクセスしよう。ポリプテルスの一番上の例を見る。
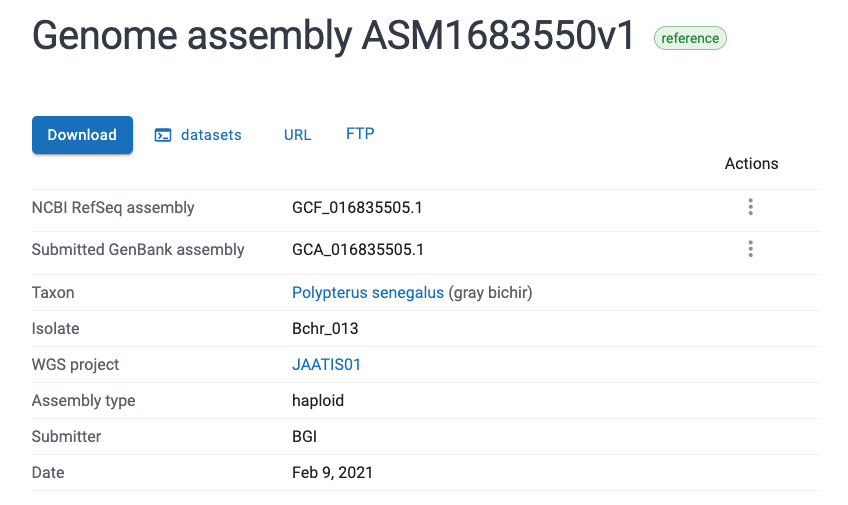
このような情報が出てくる。なるほど、2021年にBGIという組織がHaploidで決定したのだな、という情報がわかる。
更にしたに行くとアセンブルの情報がわかる。
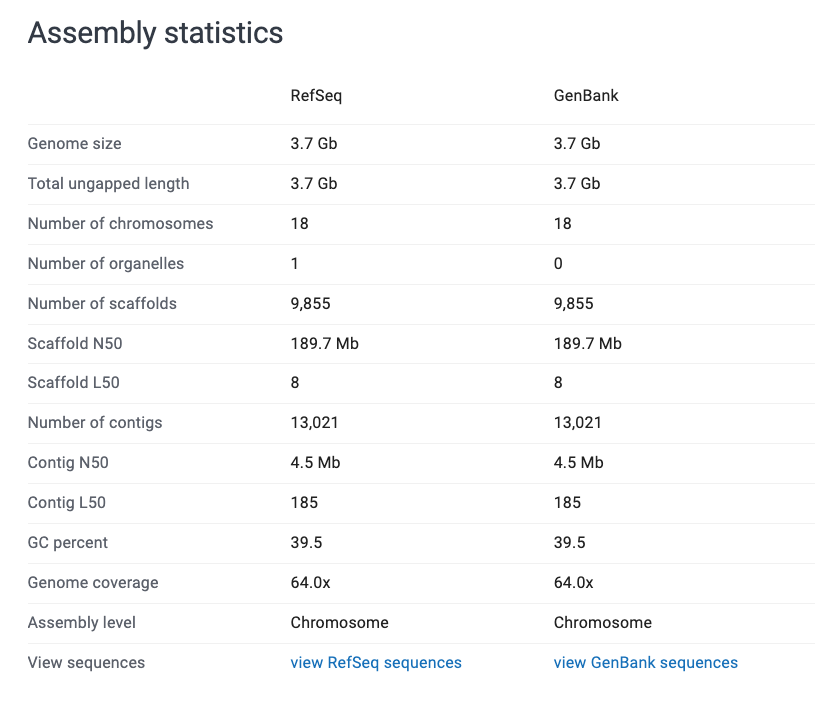
このポリプテルスのゲノムは染色体レベルで組み上がっているため、 Number of chromosomes という欄に18と書かれている。先行研究の文献を確認して核型が同じであるかも一応確認しておこう。
そして次にアセンブルの品質を見る指標として Scaffold N50 と Scaffold L50 というものがある。Scaffoldというものはコンティグ(Contig)を元に組み上げた配列のことで、一昔までは染色体レベルでアセンブルする技術がなかったので大体ここの指標で比較していた。それぞれの説明と特性は以下の通り。
| 指標 | N50 | L50 |
| 意味 | アセンブル内のすべてのScaffold を長い順に並べたとき、全Scaffoldの合計長の50%をカバーするScaffoldの長さ | アセンブル内のすべての Scaffold を長い順に並べたとき、全Scaffoldの合計長の50%をカバーするScaffoldの数 |
| 単位 | b (ベース、塩基長) | 本数 (整数値) |
| 解釈 | 値が高いほうが良い | 値が低いほうが良い |
特に近縁種でもゲノム決定がされていたり、別のバージョンのゲノムとどちらが品質が良いのだろう? というときの指標の一つとしてこれらが使える。
今回は近縁種のアミメウナギと比較してみよう。
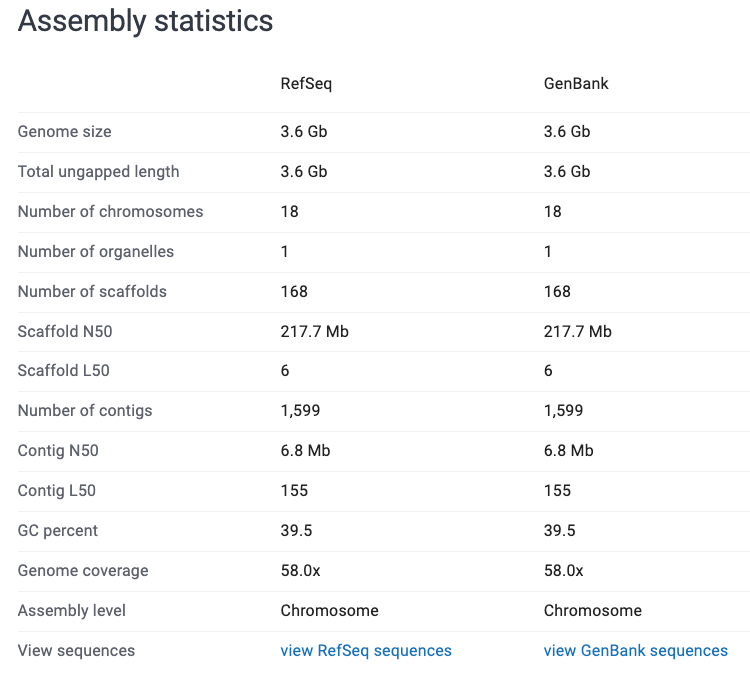
Scaffold N50に注目して見ると、アミメウナギのほうが値が高い(≒品質が高い)ことがわかる。ポリプテルスの方は染色体で組み上げたことになっているが、「行方不明のScaffold」が1万個近くあることがわかる。
あとは Genome coverage で何x(かけ)読んだか、くらいは見ると良い。エラー率に関わってくる。これは単純に読んだリードの総長をゲノムサイズで割ったもので、要は「ゲノムサイズの何倍の量のシーケンスをしたか?」ということを示す。
これのカバレッジが低いと、ギャップやエラーが出現しやすい。適切なカバレッジは20x〜50xくらいであると言われている。あまりにも低いものは少し疑ったほうがいいかもしれない。ちなみに先程のニセモノのゲノムはこんな感じ。
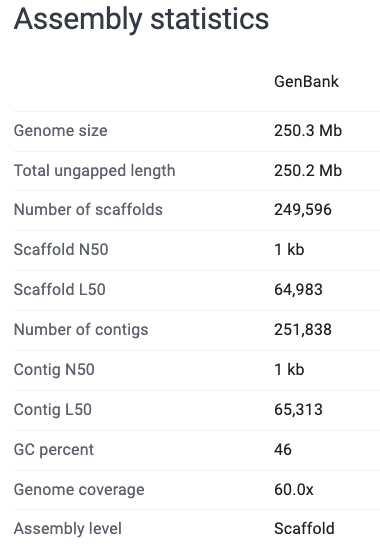
Scaffold N50が 1kb! Contig N50も 1kb ? それでも60x読んでいる……? あれれ〜おかしいよ〜。
アノテーションの情報を見る
最後にゲノムアノテーションを見てみよう。
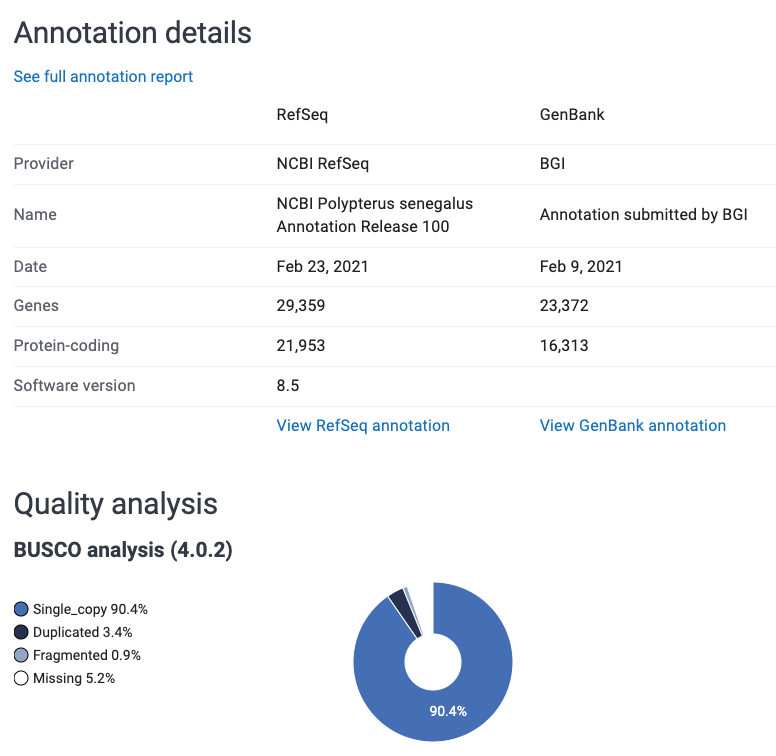
このポリプテルスの例ではGenBankには登録者が作ったアノテーション、RefSeqはNCBIが作製したパイプラインでアノテーションしたものが載っている。こちらのほうが遺伝子数が多い。
また、BUSCOについても記載されるようになった。これはゲノムの完全性を見るのに役立つツールである。
BUSCOは特定の系統群(ここではactinopterygii_odb10、つまり条鰭類)に共通する単一コピーの保存されたオーソログ遺伝子(以下BUSCO遺伝子)をもとに品質を評価している。以下の情報から、3640個の遺伝子がBUSCO遺伝子だとわかる。
C:93.8%[S:90.4%,D:3.4%],F:0.9%,M:5.2%,n:3640
actinopterygii_odb10 (3640)
という情報が書かれているので、これを見る。
まず、Cはアセンブリ内に完全なBUSCO遺伝子が検出された割合を示している。これは[S: シングルコピー、D: 完全だが複製されて検出されたBUSCO遺伝子の割合]に分けられる。Cの割合が高ければゲノムの品質は高いといえるだろう。
FはFlagmented、つまり部分的に検出されたBUSCO遺伝子の割合、MはMissing、検出されなかった割合を表している。
ただし、これらの情報はNCBIによってアノテーションがなされたゲノムでしか利用できないことに留意したい。
ダウンロード
リファレンスゲノムをダウンロードする方法。
一番上にある「Download」というところから欲しい情報にチェックを入れてダウンロードすればいい。

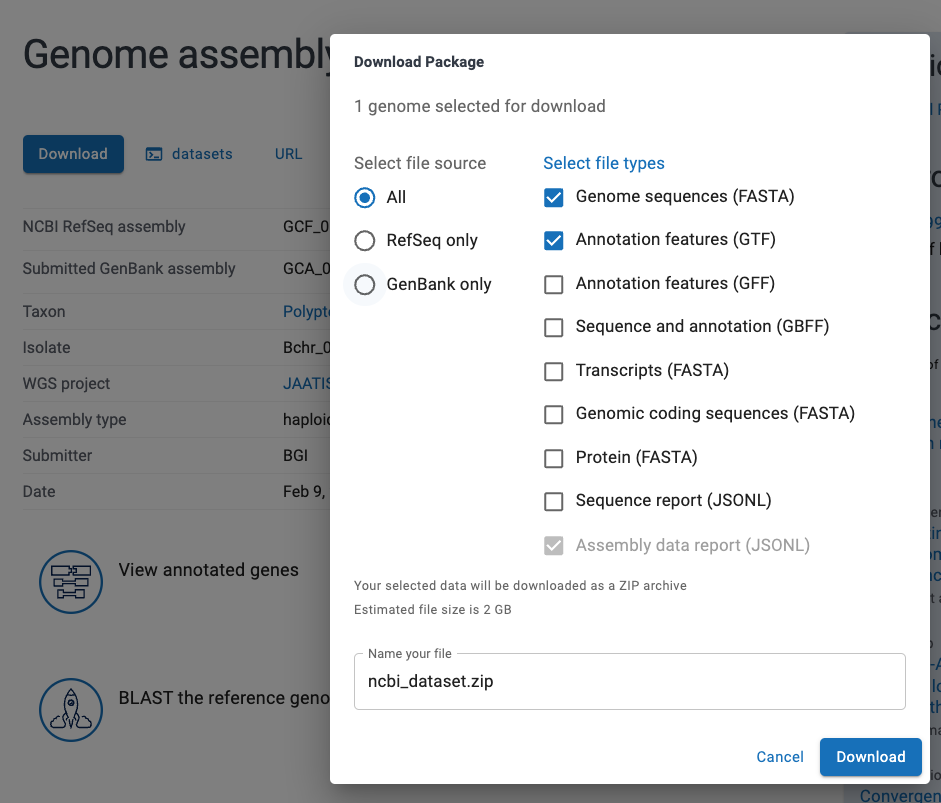
もし保存先がブラウザを操作しているPCではない場合、FTPをクリックする。
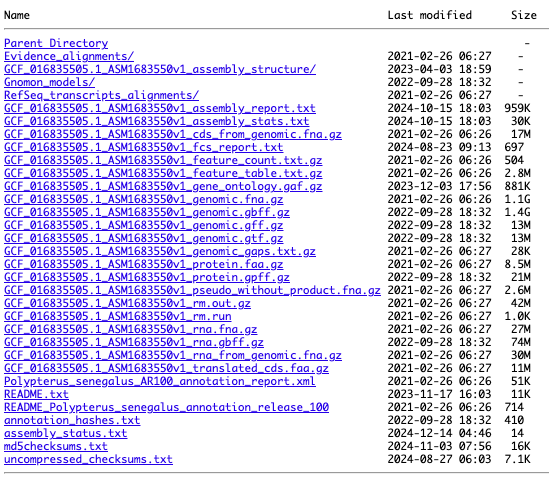
このような画面が出てくるので、必要なファイルのリンクを右クリック。
ここでChromeであればリンクのアドレスをコピーと出てくるので、それをコピーして、ターミナル上でwgetでダウンロードして使えば良い。
さいごに
2024年に大型アップデートが来て色々使い勝手が変わったが、分類群に基づいた検索ができるようになったりと色々便利な機能が盛り込まれた。
くどいようだが、生物種の背景の情報をよく理解したうえで使いこなしていこう。





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)

