
2024年11月16日 追記: 本論文は Science に掲載されました。OAではなく筆者は読めないので、最新の内容を知りたい方は本文を参照してください。また、本記事はメジャーリビジョンの前のbioRxivのv3に基づいて執筆しています。最近公開されたv4では図が新しくなっているので、もしアクセス権がないような方はこちらから論文を参照ください。(私は、科学は研究者だけのものではなく、社会全体に対して平等に知識を得る機会をもつべきだと考えています。研究者の皆様はOA推進・プレプリント投稿・機関リポジトリなどにご協力をお願いします)
昨今の基盤モデル(Foundation model)の発展は著しい。OpenAIのGPTを筆頭にして世界各国様々な企業や組織が基盤モデルへの投資を加速させている。
非機械学習の研究者においてもChatGPTを始め、CopilotやGeminiなど身近なところでも利用できる、基盤モデルをもとにしたアプリケーションを実際に研究や開発に取り入れている人も少なくないのではないかと思う。
GPTなどを筆頭にTransformerと呼ばれるアーキテクチャをベースとしたモデルが数多く登場した。ゲノムをはじめとした塩基配列や、アミノ酸配列も文字列であることから応用先として研究が進んでいる。少し前までLSTMなどの自然言語処理技術(NLP)で文字列を処理する形のものが研究されていたが、最近は基盤モデルが主流となっている。
ただその多くはアミノ酸配列、DNA、RNAといった特定の配列に焦点を当てたモデルであった。もちろん、生命はそうした一つの側面からのみ語ることができるものではなく、転写調節領域などを含むゲノム(全塩基配列)をどう扱うかが大きな課題であった。
例えば、2023年の頭に発表されたNucleotide Transformer (Dalla-Torre et al., 2023) は2.5B(25億)のパラメータで事前学習され、モデル生物・非モデル生物含めた850のゲノムからの情報を統合している。名前にあるようにTransformerをベースとしたアーキテクチャで、驚くべきことにエンハンサーなどの遺伝子発現を制御する領域を予測できたという。
さらに突然変異が遺伝子の機能に与える影響を評価することも可能であるとしている。
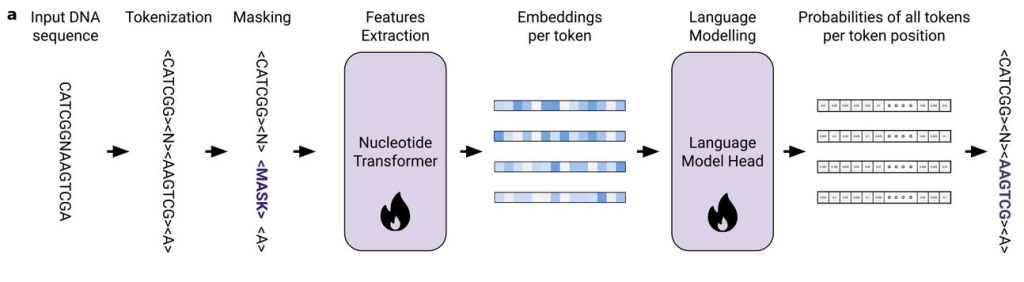
今回紹介する新しい基盤モデル Evo (Nguyen et al., 2024) はそうした流れの中で出てきたモデルである。
Evo の概要
Evoについてざっと抑えておくと、Transformerとは異なるアーキテクチャ、StripedHyena を採用し、131 kbのコンテクスト長で7B(70億)のパラメータで事前学習したモデルである。このモデルの特徴は原核生物特化型であり、なんと270万の原核生物とファージのゲノムでトレーニングされている。さらに一塩基レベルの解像度で学習されていることから、一塩基レベルの変異が及ぼす影響を予測できる。また、最大 650 kb の長さのコーディング領域を含む「ゲノム丸ごと」生成することが可能である。これまでのモデルよりも桁違いに長いゲノムを生成することができる。
モデルと学習について
さて、このモデルは具体的にどのような設計なのか見ていくと、まずStripedHyenaアーキテクチャを採用している。

StripedHyenaは長期コンテクストにおいてTransformerよりも優れたパフォーマンスを発揮することがベンチマークで報告されている。
また、面白いことに一塩基単位をトークンとしている。つまり、ATGCの4つのみの区切りで全て学習しているということだ。自然言語処理においては、通常、トークナイザと呼ばれるもので文章を単位(トークン)ごとに区切って学習させる。

従来のモデル、例えば上の Nucleotide Transformer でもある程度の長さのトークンを使用していた。ある程度まとまった配列の単位で学習させることがほとんどだ。今回このモデル、Evoでは一塩基を1トークンとしている。それを最大 131 kb のコンテクストで学習させている。
そして、他の基盤モデルと同様に生のゲノム配列に対して、次のトークンを予測するといったことを介してトレーニングしている。
ゼロショット機能予測
Evoはタンパク質コード配列を含むゲノム配列で学習されているが、本当にタンパク質の「言語」を学習していて、機能予測ができるのかどうかが焦点となる。
大腸菌タンパク質のデータセットを用いてタンパク質の機能に対する点突然変異の影響を予測するゼロショット能力を評価したところ、既存のGenSLM(コドンをもつコーディング領域でのみ学習したモデル、Zvyagin et al., 2023)よりも優れた性能を発揮した。

また、tRNAやrRNAなどのncRNAに関する機能の予測においても既存のモデルを上回っていた。また転写調節領域についても予測できるようで、これについても既存のモデルを上回っていた。
CRISPR-Cas複合体についても生成することができ、配列・構造の両方の観点から天然のCasシステムに似たものを生成することができる。
他にも、必須遺伝子の予測などにおいて Nucleotide Transformer の性能を上回っているなど、全体的に既存のモデルを上回る性能を出している。もちろん、著者らのベンチマークの結果なので、注意して見る必要がある。
ゲノムを丸ごと生成
そして、この基盤モデルで面白いのが、ゲノム丸ごと生成することができるというものだ。事前学習のコンテクストは 131 kb であるが、その約5倍の長さに相当する 650 kb 程度のゲノムを20ほどサンプリングした。その結果、Evo で生成されたゲノムにおける遺伝子領域(コーディング領域)の密度は、平均して天然のゲノムに見られる密度とほぼ同じ(下図B,C)であることが明らかになった。また、原核生物に特徴的であるように(オペロン)、隣接する遺伝子配列は同じDNA鎖(転写方向が同じ)で生成されていた。
これらの遺伝子について立体構造予測をしたところ、ほぼ全てで二次構造と球状のフォールディングが見られた(下図D, E)。一部のタンパク質については遺伝子オントロジー(GO)で注釈が付けられた既知の分子機能に関与する天然のタンパク質と構造類似性を持っていた(ただし構造予測の多くは信頼性が低いため、真に機能するかは大いに疑問)。

ただ、rRNAが含まれていないなど、生命に必須である遺伝子のいくつかは欠けているようだ。
基盤モデルがゲノムをデザインする日は近いか
今回、Evoは原核生物でのみ学習したように、複雑度が段違いの真核生物のゲノムにおいてこれがそのまま適用できるかというと正直まだまだだろう。原核生物においても、細かく見ていくと粗があるが、全体像としては天然のゲノムに近いものが生成されるようになった。
ただ、本当にここ数年で基盤モデルが大きく発展しているため、今後数年で真核生物においても同じようにゲノムレベルで生成できるような基盤モデルが登場するかもしれない。
例えばアホロートルやハイギョなどのゲノムは20〜40 Gbと非常に巨大だが、彼らは我々にはない特別な臓器・四肢再生能力を持っている。そうしたゲノムの特徴を掴み、ゲノムを「デザイン」することができるようになれば、生命科学において大きな進展が望めるだろう。それにはより長いゲノム(配列)を生成できるようなモデルの研究がキーとなるだろう。
参考文献
- Dalla-Torre, H., Gonzalez, L., Mendoza-Revilla, J., Carranza, N. L., Grzywaczewski, A. H., Oteri, F., … & Pierrot, T. (2023). The nucleotide transformer: Building and evaluating robust foundation models for human genomics. bioRxiv, 2023-01.
- Nguyen, E., Poli, M., Durrant, M. G., Thomas, A. W., Kang, B., Sullivan, J., … & Hie, B. L. (2024). Sequence modeling and design from molecular to genome scale with Evo. bioRxiv, 2024-02.
- Poli, M., Massaroli, S., Nguyen, E., Fu, D. Y., Dao, T., Baccus, S., … & Ré, C. (2023, July). Hyena hierarchy: Towards larger convolutional language models. In International Conference on Machine Learning (pp. 28043-28078). PMLR.
- 築地俊平, 新納浩幸 (2021) Tokenizerの違いによる日本語BERTモデルの性能評価 第27回言語処理学会年次大会
- Zvyagin, M., Brace, A., Hippe, K., Deng, Y., Zhang, B., Bohorquez, C. O., … & Ramanathan, A. (2023). GenSLMs: Genome-scale language models reveal SARS-CoV-2 evolutionary dynamics. The International Journal of High Performance Computing Applications, 37(6), 683-705.





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)


このような素晴らしい記事を日本語で書いていただきありがとうございます!