
現在在宅で研究をせざるを得ない状況下からか、このサイトのアクセス数が伸びている。
これを期にDRY解析を始めようと思っている人は、配列を自分で持っておらず、公開データベースを用いることが多くなると思うが、いちいち手動でFASTAファイルをダウンロードする方法はいささか効率が悪い。そこで今回は著者が開発したPythonのソフトウエアを用いて、塩基配列・アミノ酸配列をNCBI GENEからFASTA形式で効率的なダウンロードを行う方法を解説する。
一括ダウンロードツールの中身については、過去の記事を読んで欲しい。
NCBI GENEID TO FASTAを動かす環境を整える
配列の自動取得を行うソフトウエア、NCBI GENEID TO FASTA (https://github.com/kim2039/NCBI_geneIDtoFASTA)を動かすための環境を整える。
本当は製作者がDockerとか使って配布してくれればいいんだけどそこまでの技術なかったようなので(プルリクしてください)、今回はローカルに動く環境を準備する。
手法は色々あるが、一番手っ取り早い方法が”Anaconda”と呼ばれるパッケージ群を入れることだ。以下のURLから各OSに併せてダウンロードし、インストールして欲しい。https://www.anaconda.com/
以下はWindowsに準拠して話を進めていくが、基本的にどれも同じである。
また、gitをインストールしておくことをおすすめする。https://git-scm.com/
非プログラミング系の人には馴染みがないかもしれないが、最近発表される多くのソフトウエアはGitHubなどを通じて公開される。ソフトウエアに修正が入ればいちいちダウンロードし直さなければならないが、Gitを使えば差分だけ得ることができるので楽ちんである。GitHubであればZipでダウンロードすることもできるが、いちいちそれをやるのも面倒くさいので、これを期にGitを入れておくことをおすすめする。(GitはGitHubとは違うので、気になる人は調べておくと良い)
NCBI Gene から欲しい遺伝子の配列の情報を入手する
NCBI geneには様々な生物種の遺伝子情報が登録されている。多くはゲノム解読とともにRNA-seqなどの発現データなどから予測されたアノテーションが記されている。一部のモデル生物については細かい配列のキュレーションまで済まされているが、今回は非モデル生物も扱う前提で記す。
まずこちらのサイトにアクセスし、検索欄に必要な遺伝子の名前や種名などを入力して検索する。
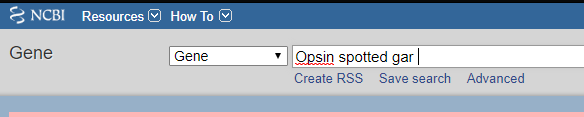
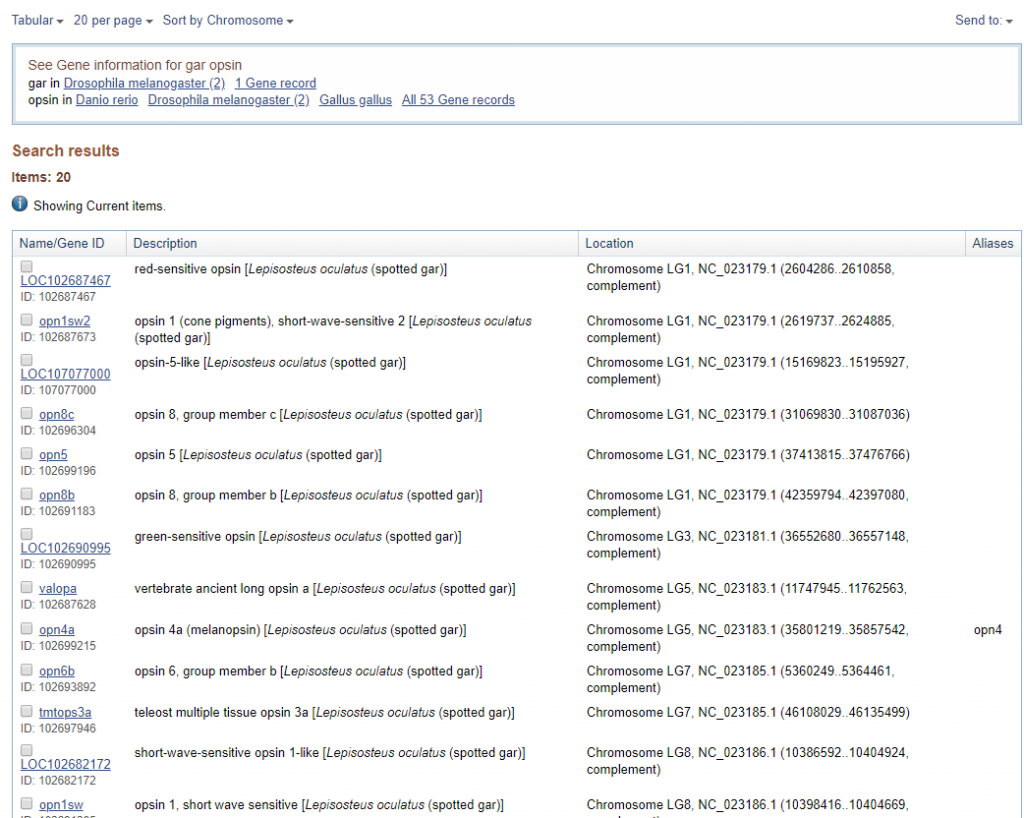
上記のように検索結果が出たら、右上にあるSend to▼ というところをクリックし、
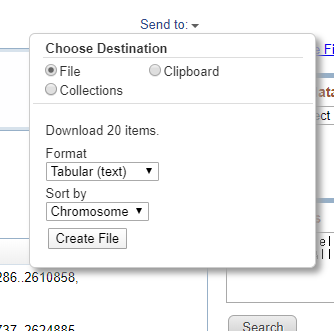
File → Tabular → Sortはお好み でCreate File をクリック
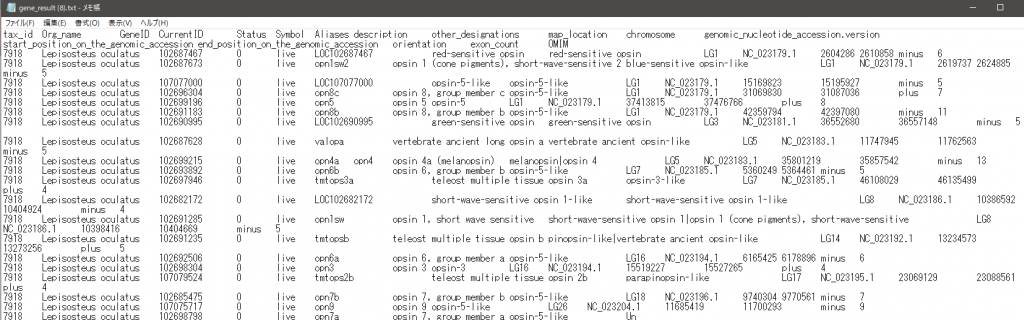
ダウンロードしたファイルを確認する。メモ帳で開くとこんな感じだが、Excelなどの表計算ソフトで開くと以下のようにTab区切りでセルに配置してくれる。
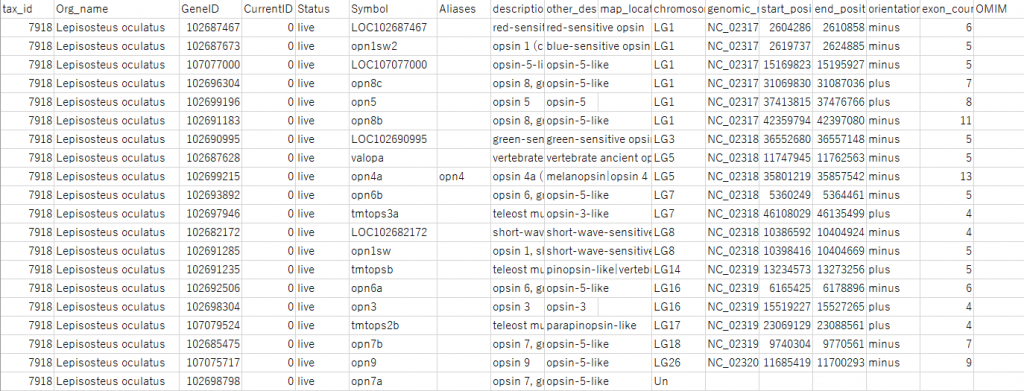
さて、自分の解析したい配列をこの中から選ぶ、もしくは関係ないものを消してしまおう。キーワード検索だけするとRelated protein などの実際に調べたい対象以外の配列が入ってしまうことが多々あるので注意である。
使う配列を決定したら、GeneIDのカラムをまるごと別のシートにコピーしてしまう。
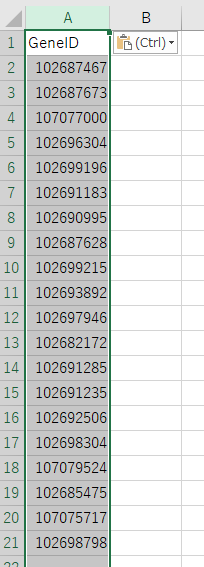
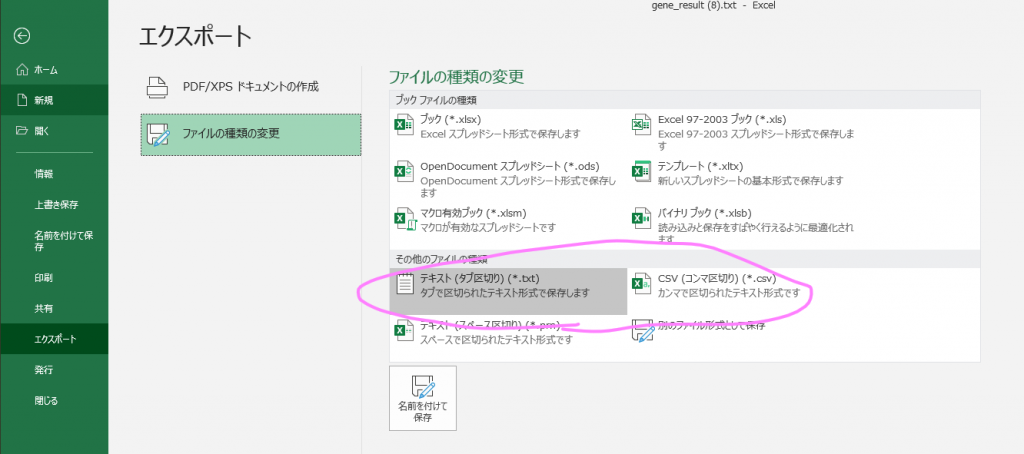
そしてこのシートだけをTSV, CSVで保存する。
NCBI geneID to FASTA をダウンロードする
ダウンロードする方法は2つほどある。Gitをダウンロード・インストールしてある人は以下のコマンドでカレントディレクトリにCloneできる。
$ git clone https://github.com/kim2039/NCBI_geneIDtoFASTA.git
もしGitを入れていないのであれば、以下の場所からZIPファイルでダウンロードできる。
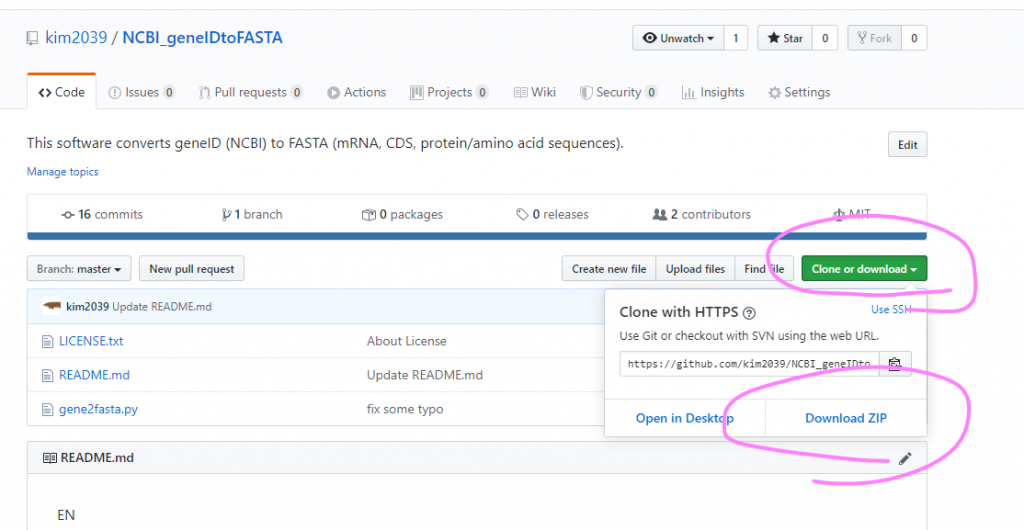
こんな感じでダウンロードできる。あとは任意のディレクトリ(フォルダ)に解凍・展開してあげればよい。
NCBI GeneID to FASTA を動かす
さて、それでは一括ダウンロードのソフトウエアを動かす。
Windowsの場合、Anaconda promptを立ち上げる。Start画面から検索してやれば出てくる。

Prompt でも Powershell でもどちらでも良い。好みがないならPowershellのほうがいい。
先程ダウンロード・Clone したディレクトリまでcdコマンドで移動する。
NCBI geneID to FASTA は以下のUsageで動作する。
$ python gene2fasta.py -h usage: gene2fasta.py [-h] [--mrna] [--protein] [--cds] [--splicing] importfile Convert geneID(NCBI) to FASTA (mRNA, CDS, protein/amino acid sequences). positional arguments: importfile TSV file input optional arguments: -h, --help このヘルプメッセージを表示する --mrna 遺伝子配列全体が必要ならばこれをつける --protein タンパク質配列が必要ならばこれをつける --cds mRNA配列をCDS配列が必要ならばこれをつける --splicing すべてのスプライシングバリアントをダウンロードするならこれをつける
といった具合である。今回はとりあえず全部盛りでダウンロードしてみよう。
$ python gene2fasta.py --mrna --protein --csd --splicing /path/to/dir/NCBIgenelist.txt
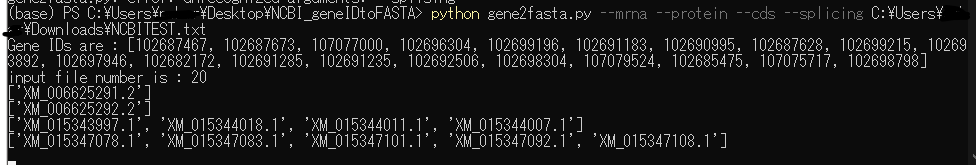
こんな感じで自動的にダウンロードしてくれる。
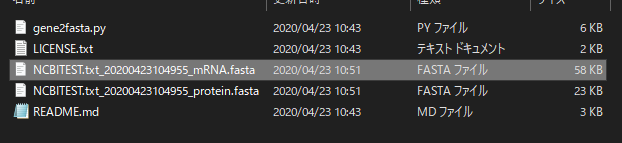
おそらくカレントディレクトリ(NCBI geneID to FASTAのファイルがあるディレクトリで実行していたならばそこ)に出力される。
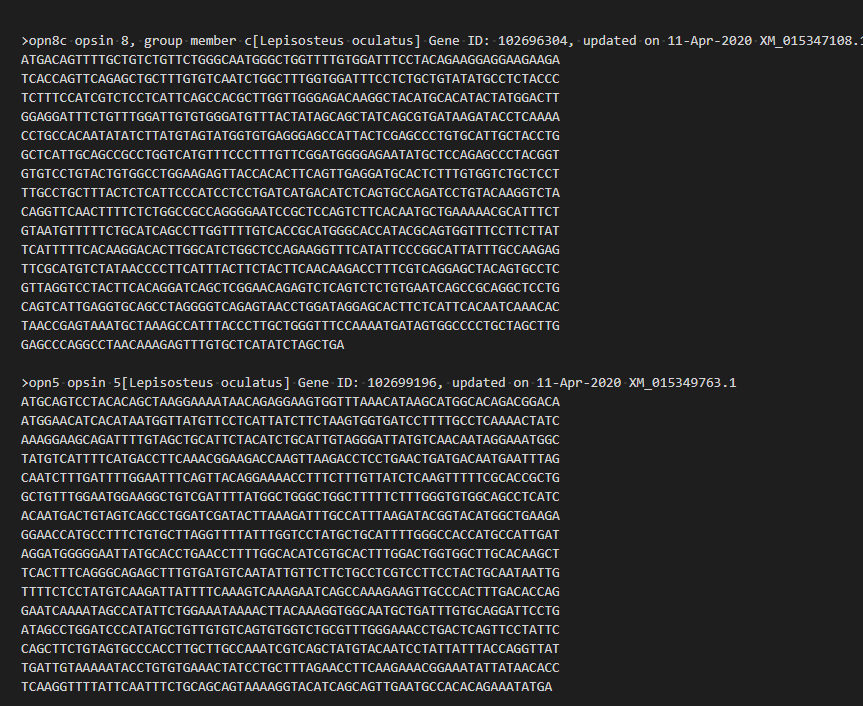
ちゃんと配列が取得できた。
さいごに
一応動作は確認しているが、バグフィックスなどが今後行われる可能性がある。
もし何かバグ等あればGitHubのissueまで。





![【Qiskit】マルチオミクス解析を量子機械学習でやる①[環境構築・基礎]](https://kimbio.info/wp-content/uploads/2024/05/2203027-100x100.jpg)

